电化学(中英文) ›› 2022, Vol. 28 ›› Issue (4): 2105181. doi: 10.13208/j.electrochem.210518
张滟滟, 刘越, 陆一鸣, 于沛平, 杜文轩, 麻冰云, 谢淼, 杨昊, 程涛*( )
)
Yan-Yan Zhang, Yue Liu, Yi-Ming Lu, Pei-Ping Yu, Wen-Xuan Du, Bing-Yun Ma, Miao Xie, Hao Yang, Tao Cheng*()
摘要:
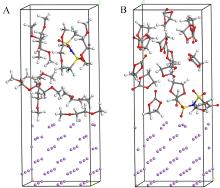
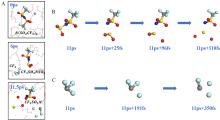
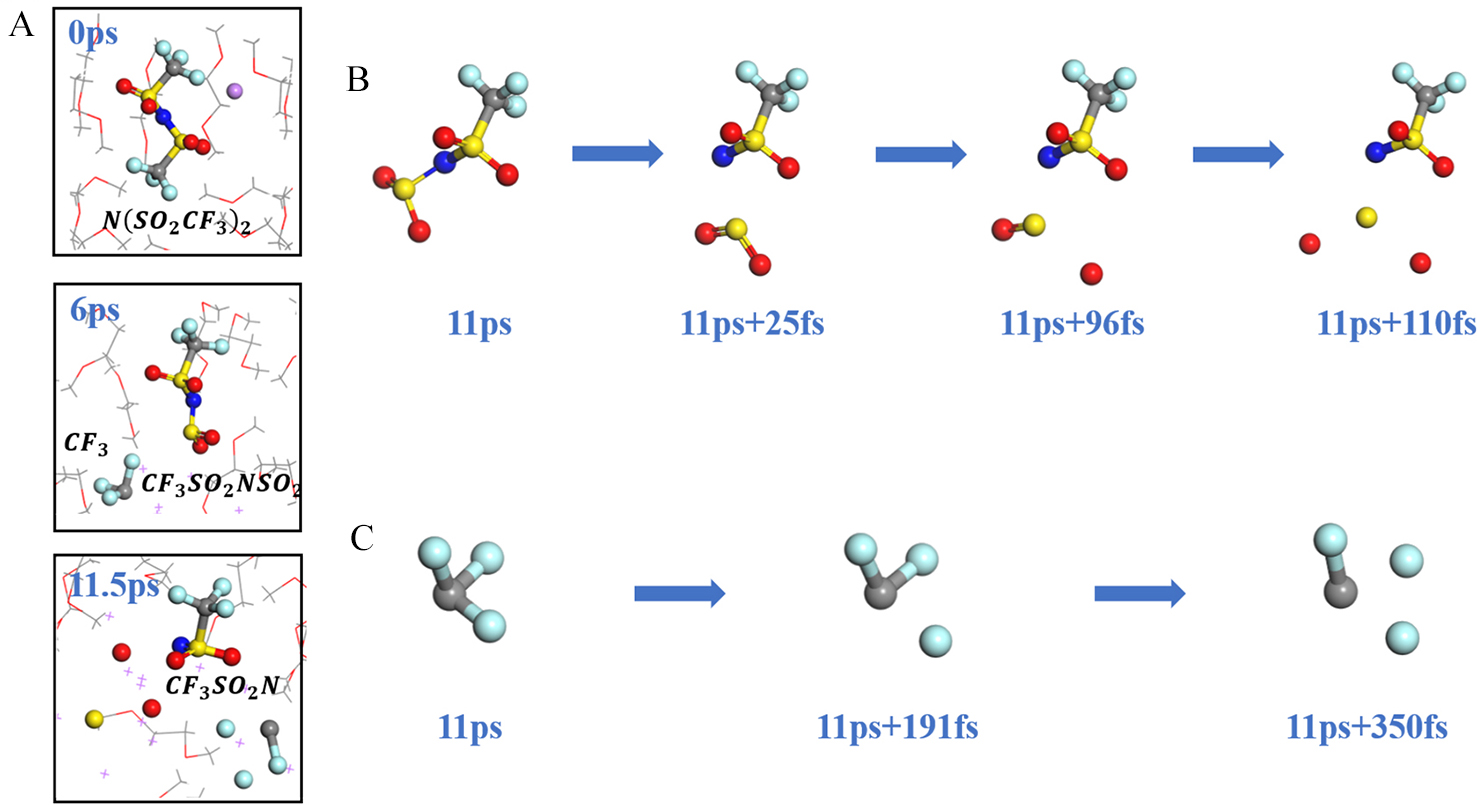
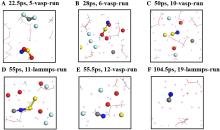
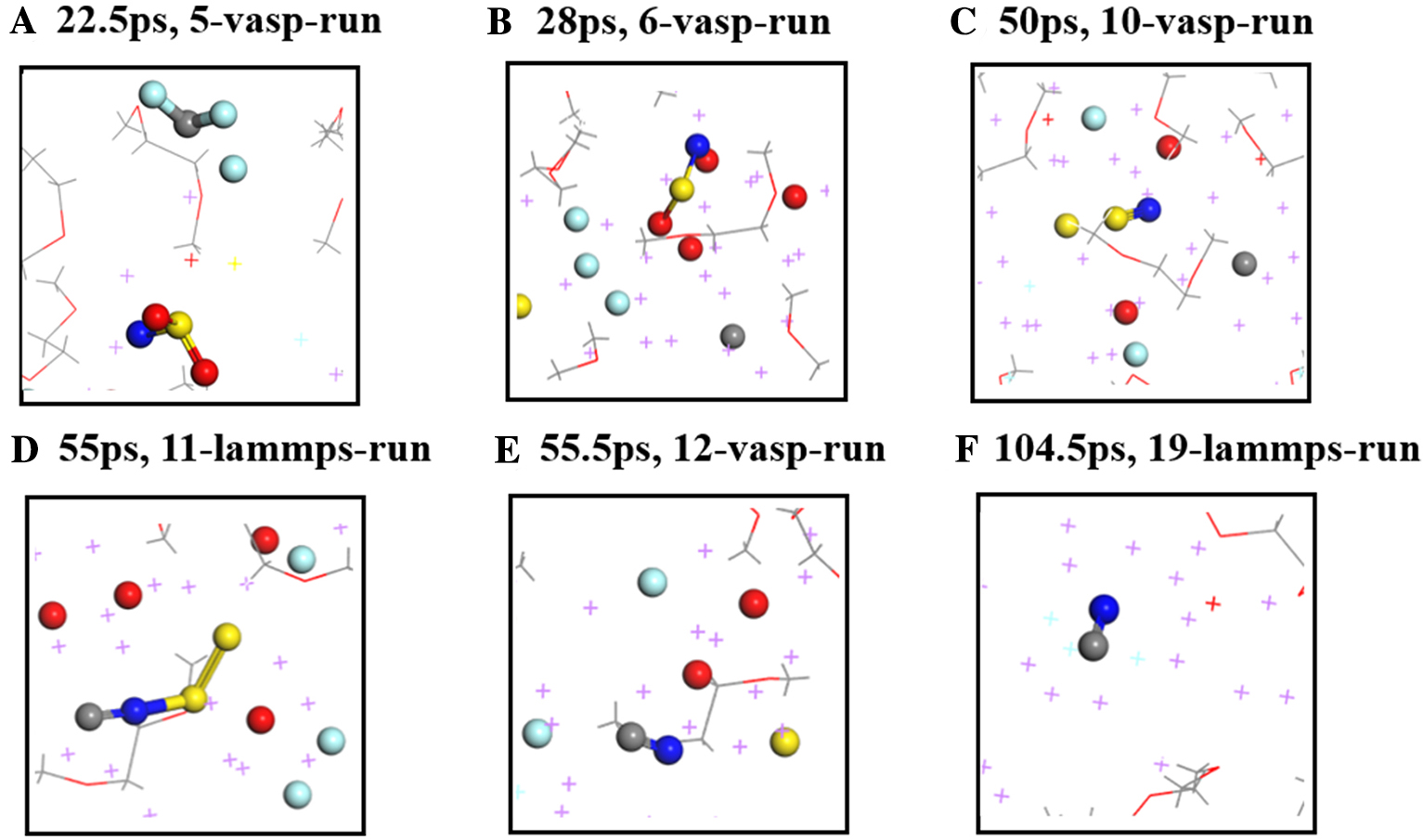
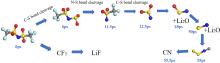
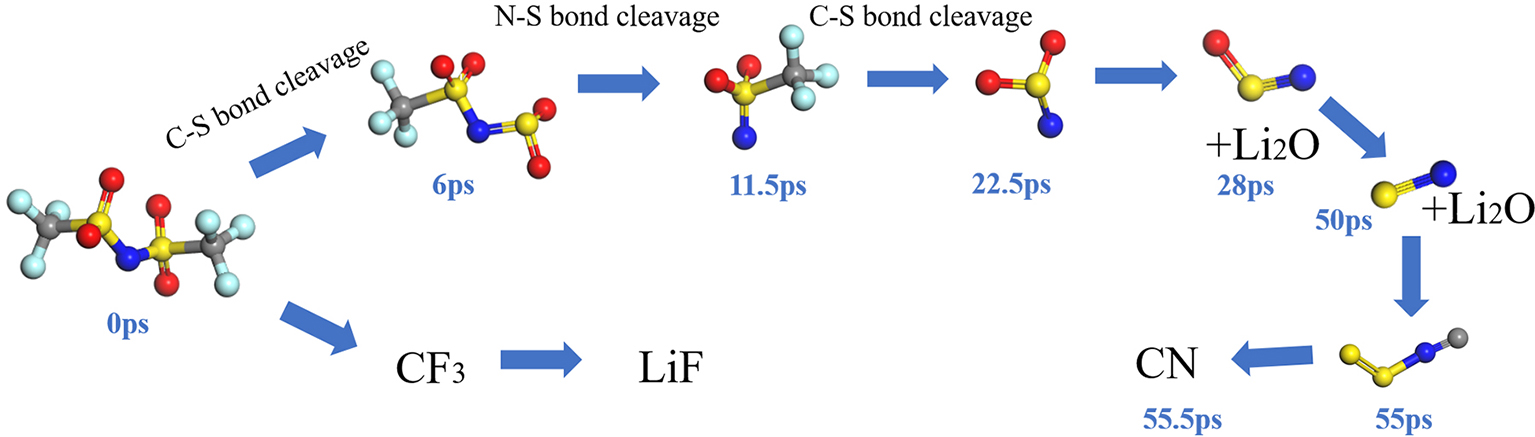
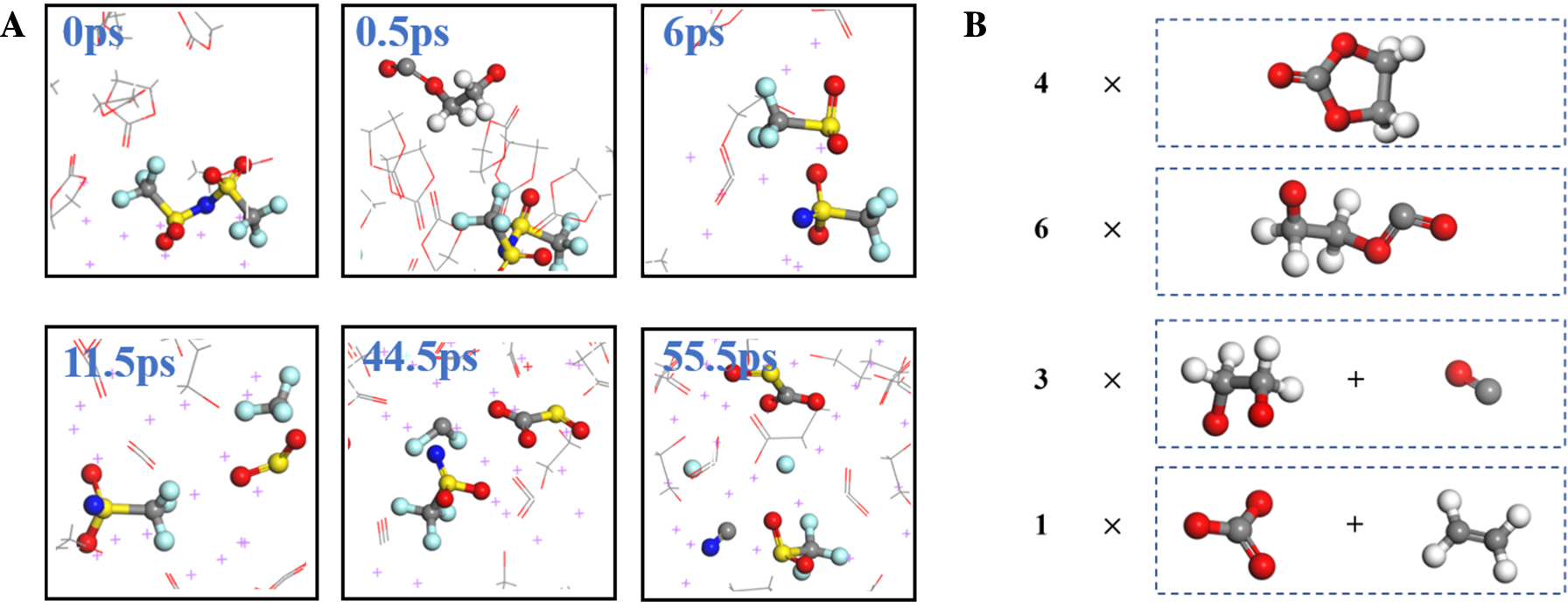
锂金属具有高比容量和极低的电极电势,被视为下一代高能量密度电池的理想负极材料。然而,锂金属具有很高的活性,在循环过程中会形成锂枝晶,刺穿固态电解质膜(solid electrolyte interphases,SEI),造成电池短路,引发一系列安全问题,上述缺点极大地阻碍了锂金属的商业应用。为了解决上述问题,理解SEI的结构及其形成原理具有重要意义。在本工作中,我们采用混合从头计算和分子动力学方法(hybrid ab initio and reactive molecule dynamics,HAIR),研究了1 mol·L-1 LiTFSI-DME(dimethoxyethane)和1 mol·L-1 LiTFSI-EC(ethylene carbonate)两种电解质溶液在锂金属表面的界面反应机理,模拟结果表明,在LiTFSI-DME电解液中,TFSI阴离子优先分解,而DME未见分解,所以TFSI起到了保护DME的作用。但是在LiTFSI-EC体系中,两者均发生了分解,说明EC稳定性较差,不利于形成稳定的SEI,上述模拟结果为通过电解质理性设计开发高性能电解质溶液体系奠定了理论基础。