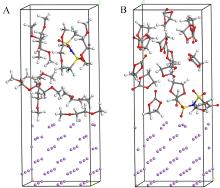
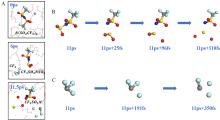
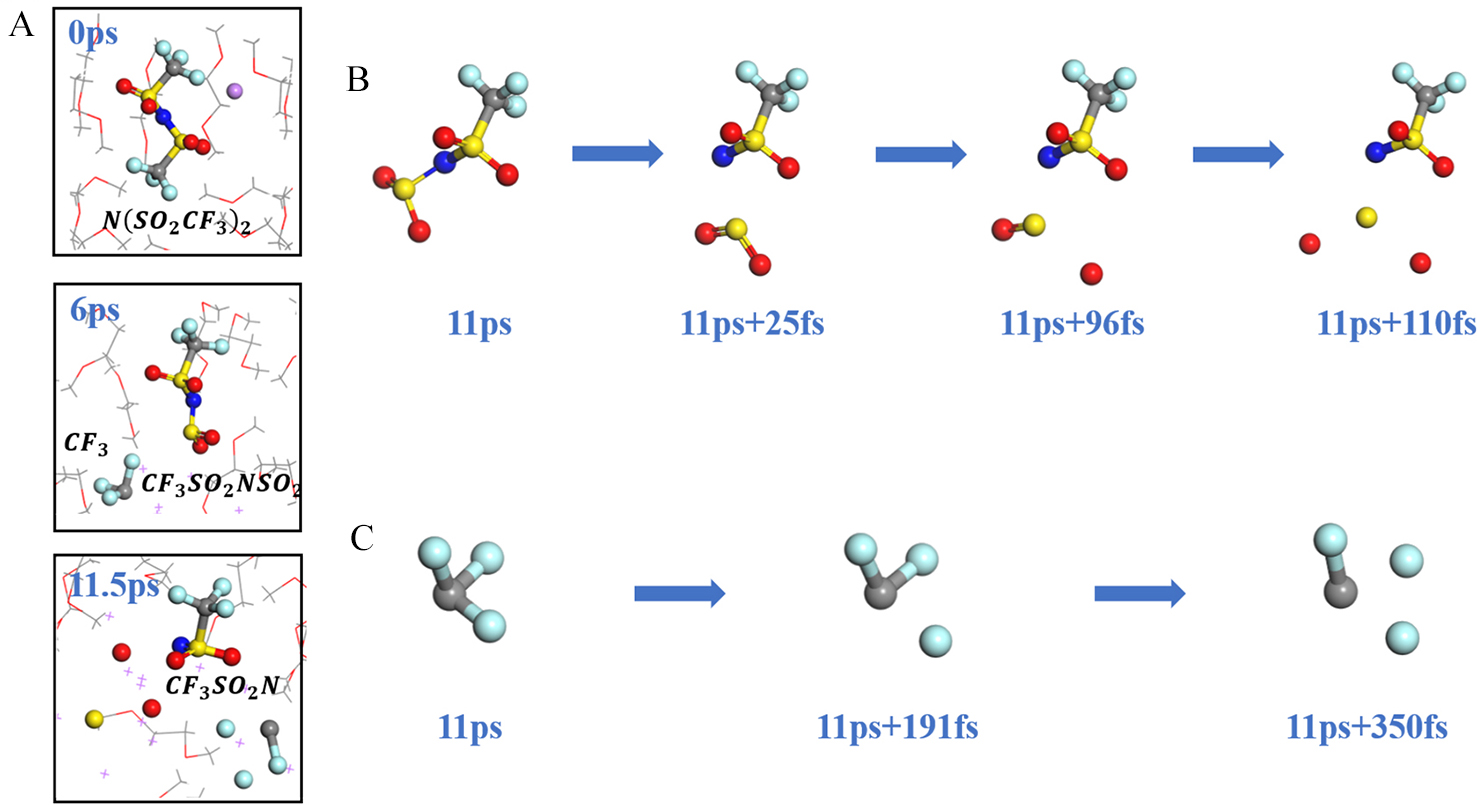
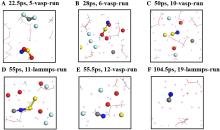
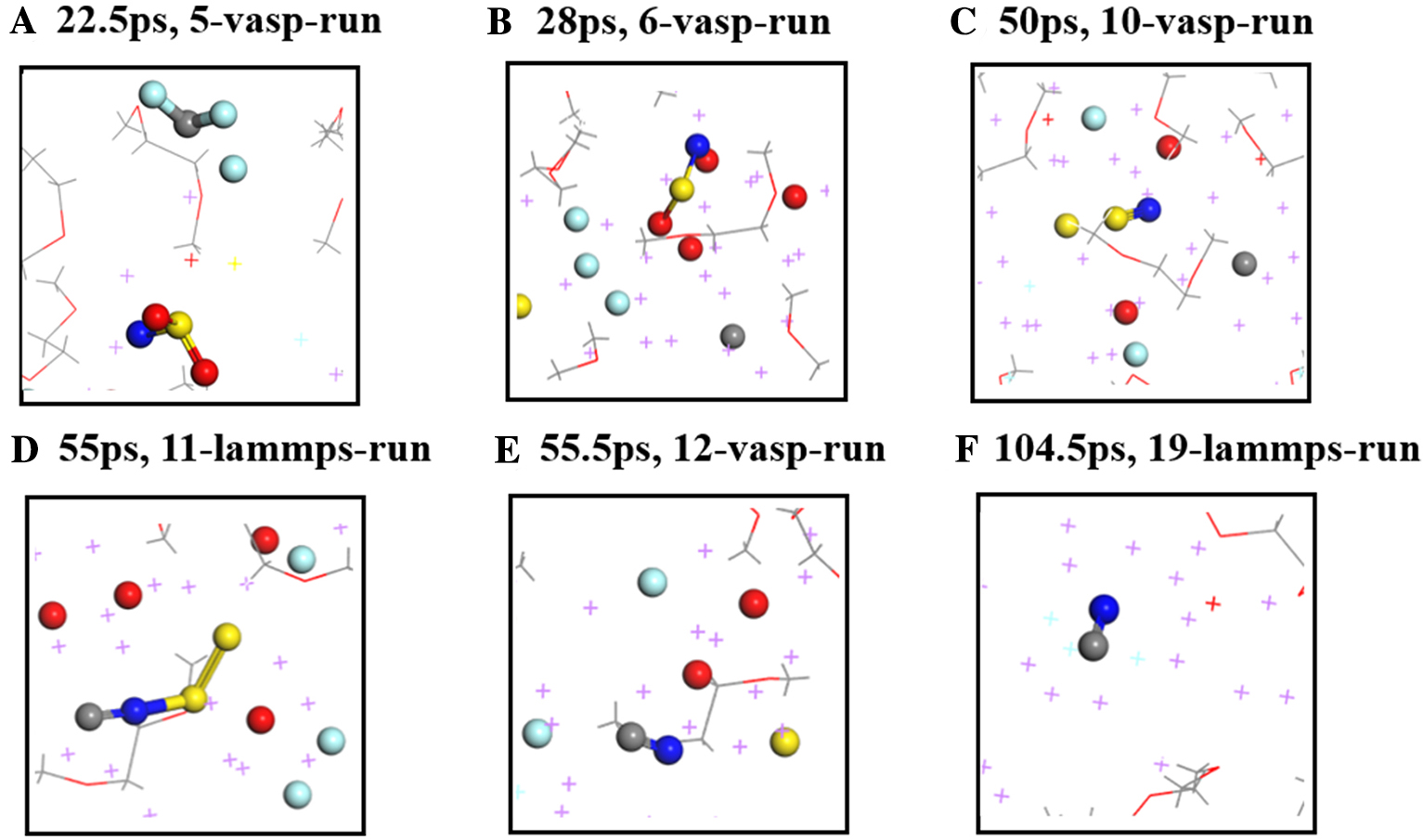
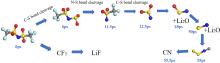
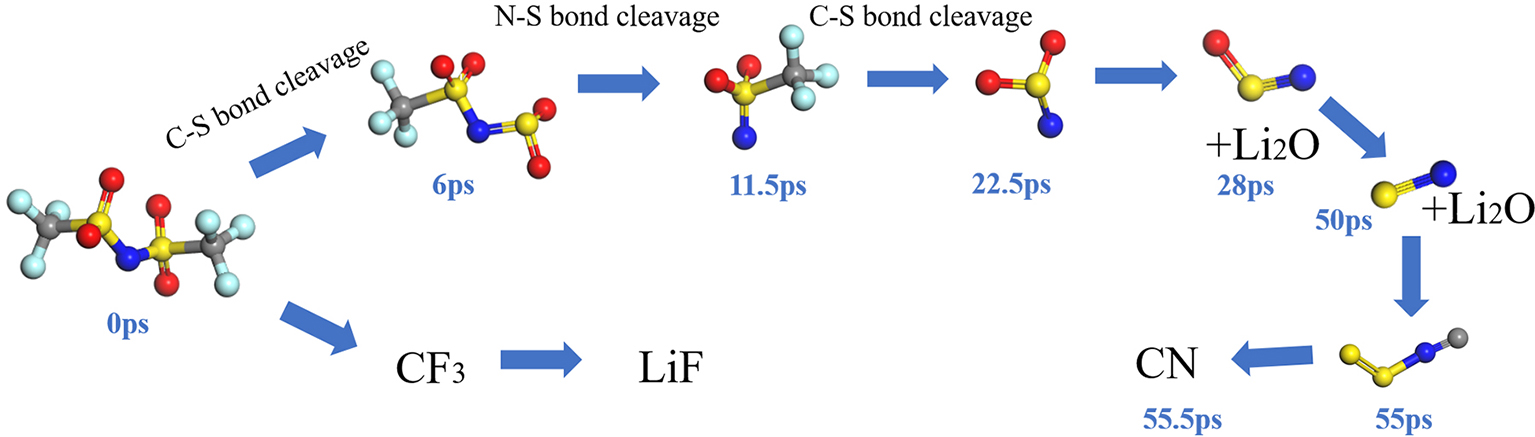
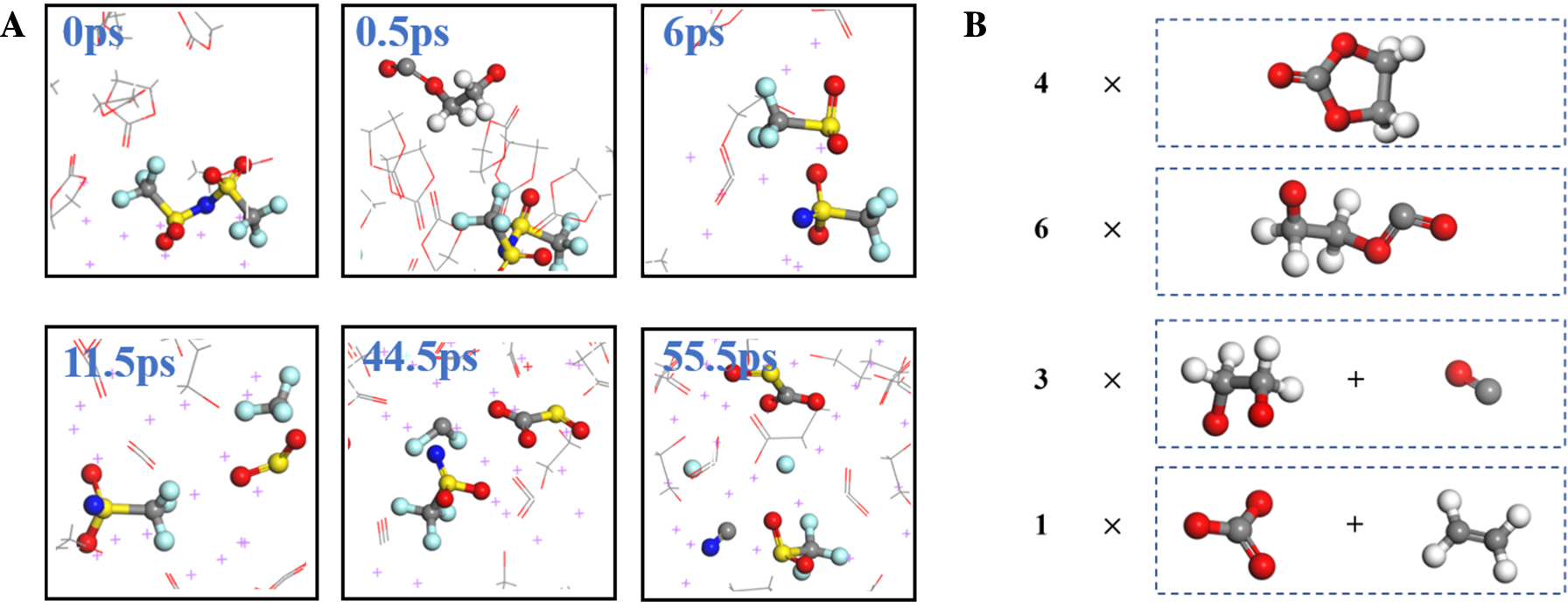
| [1] |
Li M, Lu J, Chen Z W, Amine K. 30 Years of lithium-ion batteries[J]. Adv. Mater., 2018, 30(33): 1800561.
doi: 10.1002/adma.201800561
URL
|
| [2] |
Tarascon J M, Armand M. Issues and challenges facing rechargeable lithium batteries[J]. Nature, 2001, 414(6861): 359-367.
doi: 10.1038/35104644
URL
|
| [3] |
Li P, Hwang J Y, Sun Y K. Nano/microstructured silicon-graphite composite anode for high-energy-density Li-ion battery[J]. ACS Nano, 2019, 13(2): 2624-2633.
|
| [4] |
Goodenough J B, Park K S. The Li-ion rechargeable battery: A perspective[J]. J. Am. Chem. Soc., 2013, 135(4): 1167-1176.
doi: 10.1021/ja3091438
pmid: 23294028
|
| [5] |
Xu W, Wang J L, Ding F, Chen X L, Nasybutin E, Zhang Y H, Zhang J G. Lithium metal anodes for rechargeable batteries[J]. Energy Environ. Sci., 2014, 7(2): 513-537.
doi: 10.1039/C3EE40795K
URL
|
| [6] |
Lin D C, Liu Y Y, Cui Y. Reviving the lithium metal anode for high-energy batteries[J]. Nat. Nanotechnol., 2017, 12(3): 194-206.
doi: 10.1038/nnano.2017.16
URL
|
| [7] |
Zheng J M, Engelhard M H, Mei D H, Jiao S H, Polzin B J, Zhang J G, Xu W. Electrolyte additive enabled fast charging and stable cycling lithium metal batteries[J]. Nat. Energy, 2017, 2(3): 17012.
doi: 10.1038/nenergy.2017.12
URL
|
| [8] |
Huang C, Xiao J, Shao Y Y, Zheng J M, Bennett W D, Lu D P, Saraf L V, Engelhard M, Ji L W, Zhang J G, Li X L, Graff G L, Liu J. Manipulating surface reactions in lithium-sulphur batteries using hybrid anode structures[J]. Nat. Commun., 2014, 5: 3015.
doi: 10.1038/ncomms4015
URL
|
| [9] |
Xie J, Lu Y C. A retrospective on lithium-ion batteries[J]. Nat. Commun, 2020, 11(1): 2499.
doi: 10.1038/s41467-020-16259-9
URL
|
| [10] |
Xu X L, Wang S J, Wang H, Hu C, Jin Y, Liu J B, Yan H. Recent progresses in the suppression method based on the growth mechanism of lithium dendrite[J]. Energy Chem., 2018, 27(2): 513-527.
|
| [11] |
Wang X F, Li Y J, Meng Y S. Cryogenic electron microscopy for characterizing and diagnosing batteries[J]. Joule, 2018, 2(11): 2225-2234.
doi: 10.1016/j.joule.2018.10.005
URL
|
| [12] |
Nandasiri M I, Camacho-Forero L E, Schwarz A M, Shut-thanandan V, Thevuthasan S, Balbuena P B, Mueller, K T, Murugesan V. In situ chemical imaging of solid-electrolyte interphase layer evolution in Li-S batteries[J]. Chem. Mater., 2017, 29(11): 4728-4737.
doi: 10.1021/acs.chemmater.7b00374
URL
|
| [13] |
Wang Y M, Liu Y C, Tu Y Q, Wang Q. Reductive decomposition of solvents and additives toward solid electrolyte interphase formation in lithium-ion battery[J]. J. Phys. Chem. C, 2020, 124(17): 9099-9108.
doi: 10.1021/acs.jpcc.9b10535
URL
|
| [14] |
van Duin A C T, Dasgupta S, Lorant F, Goddard W A. ReaxFF: A reactive force field for hydrocarbons[J]. J. Phys. Chem. A, 2001, 105(41): 9396-9409.
doi: 10.1021/jp004368u
URL
|
| [15] |
Chenoweth K, van Duin A C T, Goddard W A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation[J]. J. Phys. Chem. A, 2008, 112(5): 1040-1053.
doi: 10.1021/jp709896w
pmid: 18197648
|
| [16] |
Bedrov D, Smith G D, van Duin A C T. Reactions of singly-reduced ethylene carbonate in lithium battery electrolytes: A molecular dynamics simulation study using the ReaxFF[J]. J. Phys. Chem. A, 2012, 116(11): 2978-2985.
doi: 10.1021/jp210345b
URL
|
| [17] |
Liu Y, Yu P P, Wu Y, Yang H, Xie M, Huai L Y, Goddard W A, Cheng T. The DFT-ReaxFF hybrid reactive dynamics method with application to the reductive decomposition reaction of the TFSI and DOL electrolyte at a lithium-metal anode surface[J]. J. Phys. Chem. Lett., 2021, 12(4): 1300-1306.
doi: 10.1021/acs.jpclett.0c03720
URL
|
| [18] |
Liu Y, Sun Q T, Yu P P, Wu Y, Xu L, Yang H, Xie M, Cheng T, Goddard W A. Effects of high and low salt concentrations in electrolytes at lithium-metal anode surfaces using DFT-ReaxFF hybrid molecular dynamics method[J]. J. Phys. Chem. Lett., 2021, 12(11): 2922-2929.
doi: 10.1021/acs.jpclett.1c00279
URL
|
| [19] |
Islam M M, Bryantsev V S, van Duin A C T. ReaxFF reactive force field simulations on the influence of Teflon on electrolyte decomposition during Li/SWCNT anode discharge in lithium-sulfur batteries[J]. J. Electrochem. Soc., 2014, 161(8): E3009-E3014.
|
| [20] |
Metropolis N, Rosenbluth A W, Rosenbluth M N, Teller A H, Teller E. Equation of state calculations by fast computing machines[J]. J. Chem. Phys., 1953, 21(6): 1087-1092.
doi: 10.1063/1.1699114
URL
|
| [21] |
Kirkpatrick S, Gelatt C D, Vecchi M P. Optimization by simulated annealing[J]. Science, 1983, 220(4598): 671-680.
pmid: 17813860
|
| [22] |
Becke A D. Density-functional thermochemistry. III. the role of exact exchange[J]. J. Chem. Phys., 1993, 98(7): 5648-5652.
doi: 10.1063/1.464913
URL
|
| [23] |
Qian J F, Henderson W A, Xu W, Bhattacharya P, Engelhard M, Borodin O, Zhang J G. High rate and stable cycling of lithium metal anode[J]. Nat. Commun., 2015, 6: 6362.
doi: 10.1038/ncomms7362
URL
|
| [24] |
Li N W, Yin Y X, Yang C P, Guo Y G. An artificial solid electrolyte interphase layer for stable lithium metal anodes[J]. Adv. Mater., 2016, 28(9): 1853-1858.
doi: 10.1002/adma.201504526
URL
|
| [25] |
Menkin S, Golodnitsky D, Peled E. Artificial solid-electrolyte interphase (SEI) for improved cycle ability and safety of lithium-ion cells for EV applications[J]. Electro-chem. Commun., 2009, 11(9): 1789-1791.
|
| [26] |
Camacho-Forero L E, Smith T W, Bertolini S, Balbuena P B. Reactivity at the lithium-metal anode surface of lithium-sulfur batteries[J]. J. Phys. Chem. C, 2015, 119(48): 26828-26839.
doi: 10.1021/acs.jpcc.5b08254
URL
|
| [27] |
Yun K S, Pai S J, Yeo B C, Lee K R, Kim S J, Han S S. Simulation protocol for prediction of a solid-electrolyte interphase on the silicon-based anodes of a lithium-ion battery: ReaxFF reactive force field[J]. J. Phys. Chem. Lett., 2017, 8(13): 2812-2818.
doi: 10.1021/acs.jpclett.7b00898
URL
|

 )
)