电化学(中英文) ›› 2023, Vol. 29 ›› Issue (10): 2205171. doi: 10.13208/j.electrochem.2205171
• 论文 • 上一篇
乔行a,#, 朱勇a,#, 孙升a,b,*( ), 张统一a,*()
), 张统一a,*()
Hang Qiaoa,#, Yong Zhua,#, Sheng Suna,b,*(), Tong-Yi Zhanga,*()
摘要:
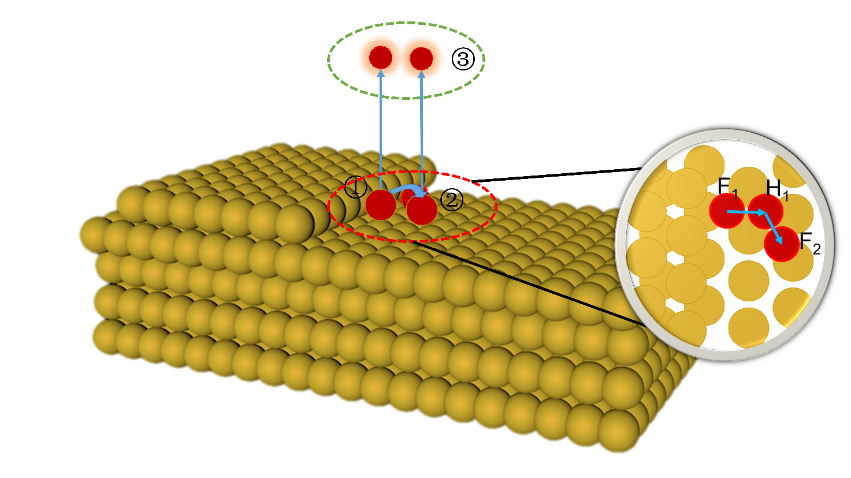
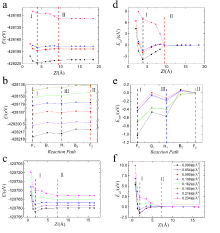
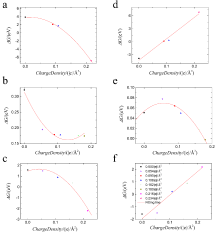
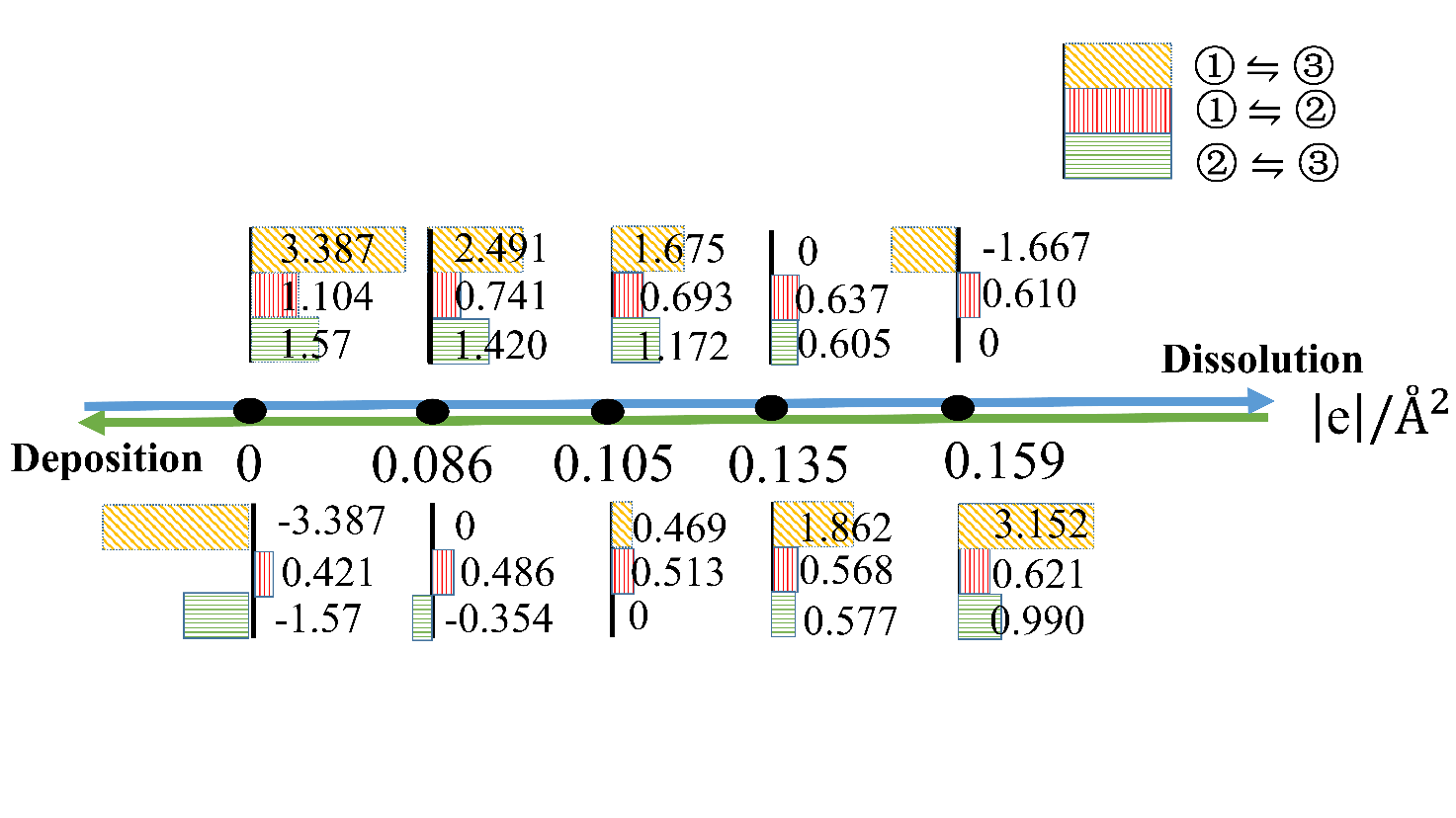
电化学沉积和电化学腐蚀的核心问题是不同电压/电荷作用下的电极/电解质界面行为,其控制量是溶解/沉积反应路径的势垒,但是势垒的测量和计算难度比较大。本文采用密度泛函和连续介质耦合方法研究了不同加载电荷面密度下平整表面和含阶梯表面的Cu(111)面薄板电极直接和间接溶解/沉积两种路径的能量形态。结果发现,不同加载电荷面密度下溶质Cu原子在Cu(111)面的表面扩散和溶解过程中初末态能量分别和最高过渡态能量存在简单的线性关系,符合经典的Brønsted-Evans-Polanyi关系。在直接/间接溶解和沉积过程中,势垒和加载的电荷面密度呈线性或二次函数关系。通过这些表达式可以直接从稳态能量计算溶解/沉积和表面扩散的势垒,也可以直接计算不同加载电荷面密度下的势垒,极大的降低实验和计算工作量。通过拟合公式计算出不同临界加载电荷面密度时的势垒大小可以得出:对于溶解过程中,随着加载电荷面密度逐渐增大至0.135 |e|/Å2,阶梯处原子首先以直接溶解的方式进入到电解质溶液中;对于沉积过程,随着加载电荷面密度降低至0.105 |e|/Å2,电沉积首先发生在平整表面,并可越过较低的表面扩散势垒移动至台阶处,表面扩散是速率控制步骤。当加载电荷面密度进一步减小为0.086 |e|/Å2,此时的沉积方式以直接沉积到阶梯位置为主。