
基于从头算分子动力学的金属/氧化物-水界面能带排列
收稿日期: 2022-06-20
修回日期: 2022-07-11
录用日期: 2022-09-02
网络出版日期: 2022-12-02
基金资助
国家自然科学基金项目(22021001);国家自然科学基金项目(21991151);国家自然科学基金项目(21991150)
Band Alignments of Metal/Oxides-Water Interfaces Using Ab Initio Molecular Dynamics
Received date: 2022-06-20
Revised date: 2022-07-11
Accepted date: 2022-09-02
Online published: 2022-12-02
庄永斌 , 程俊 . 基于从头算分子动力学的金属/氧化物-水界面能带排列[J]. 电化学, 2023 , 29(7) : 2216001 . DOI: 10.13208/j.electrochem.2216001
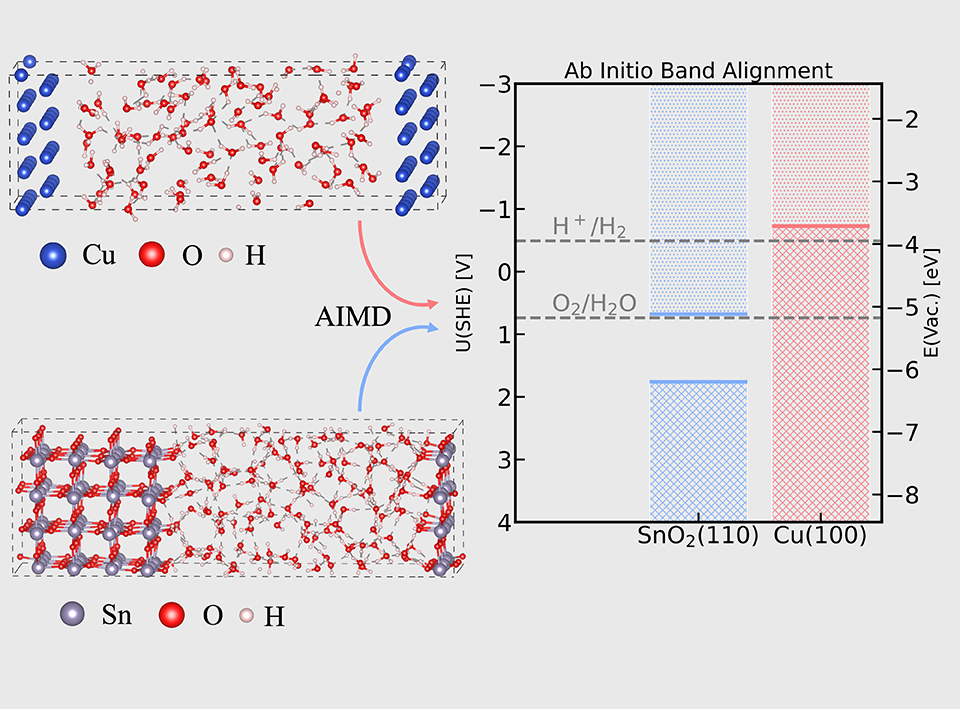
Band alignments of electrode-water interfaces are of crucial importance for understanding electrochemical interfaces. In the scenario of electrocatalysis, applied potentials are equivalent to the Fermi levels of metals in the electrochemical cells; in the scenario of photo(electro)catalysis, semiconducting oxides under illumination have chemical reactivities toward redox reactions if the redox potentials of the reactions straddle the conduction band minimums (CBMs) or valence band maximums (VBMs) of the oxides. Computational band alignments allow us to obtain the Fermi level of metals, as well as the CBM and VBM of semiconducting oxides with respect to reference electrodes. In this tutorial, we describe how to obtain the band alignments using ab initio molecular dynamics simulations. To be simple, we introduce the protocol of computational band alignments through two selected charge-neutral interfaces, i.e., Cu(100)- and SnO2(110)-water interfaces. It should be bear in mind that one can also apply this protocol to electrified interfaces. The band alignments at charge-neutral interfaces have different meanings for metals and semiconducting oxides. For metals, the alignments amount to Potentials of Zero Charge of metals, under which the metal-water interfaces possess zero net charge. For semiconducting oxides, the alignments show the positions of CBMs and VBMs under a special pH and potential. The special pH is named as Point of Zero Charge and the special potential is called Flat-Band Potential. The oxides-water interfaces have zero net charge if they are at the special pH and potential. It is worth noting that neither the positions of CBMs nor VBMs are directly interpreted as applied potentials. In the protocol, we refer computed levels to standard hydrogen electrode (SHE), and thus directly compare the levels with those from electrochemical experiments. With PBE functional, the computed Fermi level of Cu(100) is -0.726 V with respect to SHE and matches the experimental determination of -0.73 V (SHE). The CBM and VBM of SnO2(110), however, are computed as 1.76 V and 0.6 V (SHE), respectively, which fails to match the experimental values of 3.747 V and 0.147 V (SHE), respectively. We attribute the failure to the delocalization error of density functional theory. Because of the error, DFT tends to spatially delocalize one-electron orbitals, which occasionally has negligible influences on the Fermi level of metal, but significantly underestimates the band gaps of semiconducting oxides.
| [1] | Sato N. Electrochemistry at metal and semiconductor electrodes[M]. America: Elsevier Science, 1998: 119-199. |
| [2] | Le J, Cuesta A, Cheng J. The structure of metal-water interface at the potential of zero charge from density functional theory-based molecular dynamics[J]. J. Electroanal. Chem., 2018, 819: 87-94. |
| [3] | Le J, Iannuzzi M, Cuesta A, Cheng J. Determining potentials of zero charge of metal electrodes versus the standard hydrogen electrode from density-functional-theory-based molecular dynamics[J]. Phys. Rev. Lett., 2017, 119(1): 016801. |
| [4] | Le J B, Chen A, Li L, Xiong J F, Lan J, Liu Y P, Iannuzzi M, Cheng J. Modeling electrified Pt(111)-had/water interfaces from ab initio molecular dynamics[J]. JACS Au, 2021, 1(5): 569-577. |
| [5] | Le J B, Fan Q Y, Li J Q, Cheng J. Molecular origin of negative component of helmholtz capacitance at electrified pt(111)/water interface[J]. Sci. Adv., 2020, 6(41): eabb1219. |
| [6] | Li C Y, Le J B, Wang Y H, Chen S, Yang Z L, Li J F, Cheng J, Tian Z Q. In situ probing electrified interfacial water structures at atomically flat surfaces[J]. Nat. Mater., 2019, 18(7): 697-701. |
| [7] | Ledezma-Yanez I, Wallace W D Z, Sebastián-Pascual P, Climent V, Feliu J M, Koper M T M. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes[J]. Nat. Energy, 2017, 2(4): 17031. |
| [8] | Memming R. Semiconductor electrochemistry, 2nd edition[M]. America: Wiley-VCH Verlag Gmbh, 2018. |
| [9] | Cheng J, Sprik M. Alignment of electronic energy levels at electrochemical interfaces[J]. Phys. Chem. Chem. Phys., 2012, 14(32): 11245-11267. |
| [10] | Jia M, Zhang C, Cheng J. Origin of asymmetric electric double layers at electrified oxide/electrolyte interfaces[J]. J. Phys. Chem. Lett., 2021, 12(19): 4616-4622. |
| [11] | Zhang C, Hutter J, Sprik M. Coupling of surface chemistry and electric double layer at TiO2 electrochemical interfaces[J]. J. Phys. Chem. Lett., 2019, 10(14): 3871-3876. |
| [12] | Cheng J, Liu X D, VandeVondele J, Sulpizi M, Sprik M. Redox potentials and acidity constants from density functional theory based molecular dynamics[J]. Accounts Chem. Res., 2014, 47(12): 3522-3529. |
| [13] | Cheng J, Sulpizi M, Sprik M. Redox potentials and pKa for benzoquinone from density functional theory based molecular dynamics[J]. J. Chem. Phys., 2009, 131(15): 154504. |
| [14] | Costanzo F, Sulpizi M, Valle R G D, Sprik M. The oxidation of tyrosine and tryptophan studied by a molecular dynamics normal hydrogen electrode[J]. J. Chem. Phys., 2011, 134(24): 244508. |
| [15] | Guo Z, Ambrosio F, Chen W, Gono P, Pasquarello A. Alignment of redox levels at semiconductor-water interfaces[J]. Chem. Mater., 2018, 30(1): 94-111. |
| [16] | Pham T A, Lee D, Schwegler E, Galli G. Interfacial effects on the band edges of functionalized Si surfaces in liquid water[J]. J. Am. Chem. Soc., 2014, 136(49): 17071-17077. |
| [17] | Cheng J, Sprik M. Aligning electronic energy levels at the TiO2/H2O interface[J]. Phys. Rev. B, 2010, 82(8): 081406. |
| [18] | Kühne T D, Iannuzzi M, Ben M D, Rybkin V V, Seewald P, Stein F, Laino T, Khaliullin R Z, Schütt O, Schiffmann F, Golze D, Wilhelm J, Chulkov S, Bani-Hashemian M H, Weber V, Bor?tnik U, Taillefumier M, Jakobovits A S, Lazzaro A, Pabst H, Müller T, Schade R, Guidon M, Andermatt S, Holmberg N, Schenter G K, Hehn A, Bussy A, Belleflamme F, Tabacchi G, Gl?? A, Lass M, Bethune I, Mundy CJ, Plessl C, Watkins M, VandeVondele J, Krack M, Hutter J. Cp2k: An electronic structure and molecular dynamics software package-quickstep: Efficient and accurate electronic structure calculations[J]. J. Chem. Phys., 2020, 152(19): 194103. |
| [19] | VandeVondele J, Hutter J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases[J]. J. Chem. Phys., 2007, 127(11): 114105. |
| [20] | Goedecker S, Teter M, Hutter J. Separable dual-space gaussian pseudopotentials[J]. Phys. Rev. B, 1995, 54(3): 1703-1710. |
| [21] | Krack M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals[J]. Theor. Chem. Acc., 2005, 114(1-3): 145-152. |
| [22] | Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Phys. Rev. Lett., 1996, 77(18): 3865-3868. |
| [23] | Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory[J]. J. Comput. Chem., 2011, 32(7): 1456-1465. |
| [24] | Nosé S. A molecular dynamics method for simulations in the canonical ensemble[J]. Mol. Phys., 1984, 52(2): 255-268. |
| [25] | Nosé S. A unified formulation of the constant temperature molecular dynamics methods[J]. J. Chem. Phys., 1984, 81(1): 511-519. |
| [26] | VandeVondele J, Mohamed F, Krack M, Hutter J, Sprik M, Parrinello M. The influence of temperature and density functional models in ab initio molecular dynamics simulation of liquid water[J]. J. Chem. Phys., 2005, 122(1): 014515. |
| [27] | Li X Y, Chen A, Yang X H, Zhu J X, Le J B, Cheng J. Linear correlation between water adsorption energies and volta potential differences for metal/water interfaces[J]. J. Phys. Chem. Lett., 2021, 12(30): 7299-7304. |
| [28] | ?ukomska A, Sobkowski J. Potential of zero charge of monocrystalline copper electrodes in perchlorate solutions[J]. J. Electroanal. Chem., 2004, 567(1): 95-102. |
| [29] | Cheng J, Sprik M. The electric double layer at a rutile TiO2 water interface modelled using density functional theory based molecular dynamics simulation[J]. J. Phys. Condens. Matter., 2014, 26(24): 244108. |
| [30] | Bogdanova N F, Klebanov A V, Ermakova L E, Sidorova M P, Aleksandrov D A. Adsorption of ions on the surface of tin dioxide and its electrokinetic characteristics in 1 : 1 electrolyte solutions[J]. Colloid. Journal., 2004, 66(4): 409-417. |
| [31] | Bandara J, Pradeep U W. Tuning of the flat-band potentials of nanocrystalline TiO2 and SnO2 particles with an outer-shell mgo layer[J]. Thin Solid Films, 2008, 517(2): 952-956. |
/
| 〈 |
|
〉 |
