
多尺度模拟研究溶质调控下电解液在锂金属电极上的分解机理
收稿日期: 2021-05-18
修回日期: 2021-08-04
网络出版日期: 2021-07-20
基金资助
国家自然科学基金项目(21903058);国家自然科学基金项目(22003044);江苏省自然科学基金(编号SBK20190810);江苏省六大人才高峰(编号JNHB-106);及江苏省高等学校优先发展的学术项目(PAPD编号)
Multi-Scale Simulation Revealing the Decomposition Mechanism of Electrolyte on Lithium Metal Electrode
Received date: 2021-05-18
Revised date: 2021-08-04
Online published: 2021-07-20
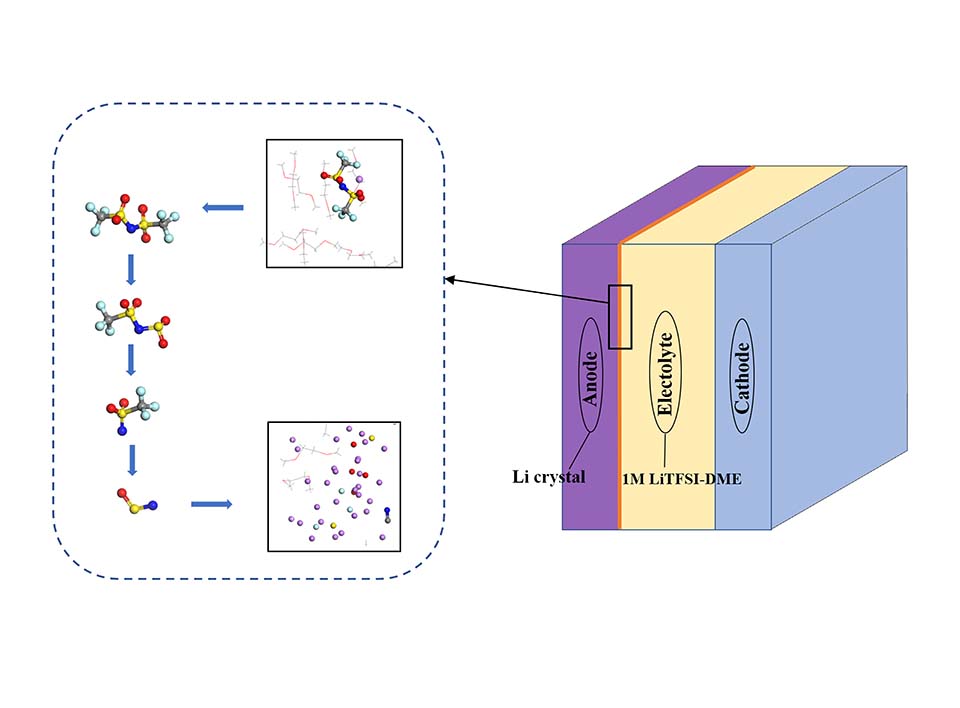
锂金属具有高比容量和极低的电极电势,被视为下一代高能量密度电池的理想负极材料。然而,锂金属具有很高的活性,在循环过程中会形成锂枝晶,刺穿固态电解质膜(solid electrolyte interphases,SEI),造成电池短路,引发一系列安全问题,上述缺点极大地阻碍了锂金属的商业应用。为了解决上述问题,理解SEI的结构及其形成原理具有重要意义。在本工作中,我们采用混合从头计算和分子动力学方法(hybrid ab initio and reactive molecule dynamics,HAIR),研究了1 mol·L-1 LiTFSI-DME(dimethoxyethane)和1 mol·L-1 LiTFSI-EC(ethylene carbonate)两种电解质溶液在锂金属表面的界面反应机理,模拟结果表明,在LiTFSI-DME电解液中,TFSI阴离子优先分解,而DME未见分解,所以TFSI起到了保护DME的作用。但是在LiTFSI-EC体系中,两者均发生了分解,说明EC稳定性较差,不利于形成稳定的SEI,上述模拟结果为通过电解质理性设计开发高性能电解质溶液体系奠定了理论基础。
张滟滟 , 刘越 , 陆一鸣 , 于沛平 , 杜文轩 , 麻冰云 , 谢淼 , 杨昊 , 程涛 . 多尺度模拟研究溶质调控下电解液在锂金属电极上的分解机理[J]. 电化学, 2022 , 28(4) : 2105181 . DOI: 10.13208/j.electrochem.210518
Lithium metal is considered as an ideal anode material for next-generation high energy density batteries with its high specific capacity and low electrode potential. However, the high activity of lithium metal can lead to a series of safety issues. For example, lithium metal will continuously react chemically with the electrolyte, forming unstable the solid electrolyte (SEI) films. In addition, lithium dendrites can be formed during cycling, which can puncture the SEI film and cause short circuits in the battery. These drawbacks greatly hinder the commercial application of lithium metal. To solve the above problems, it is important to understand the structure of SEI and the underlying mechanism of its formation as a guide for rational design. Quantum mechanics (QM) has been demonstrated as an effective tool to investigate the chemical reactions and microscopic atomic structures of SEI. However, QM is computationally too expensive to be used for large-scale and long-term theoretical simulations. Instead, the molecular mechanics (MM) method has much orders higher computational efficiency than QM, and can be used for large-scale and long-time theoretical simulations. However, the accuracy of MM is usually not guaranteed, especially for complex SEI. Therefore, a practical solution is to combine the advantages of both. In this work, we use the hybrid ab initio and reactive molecule dynamics (HAIRs) approach to describe chemical reactions with the accuracy of quantum chemistry and improve the computational efficiency by more than 10 times with mixing QM and MM. Using this method, we have investigated the interfacial reaction mechanism of two electrolyte solutions, 1 mol·L-1 LiTFSI-DME (dimethoxyethane) and 1 mol·L-1 LiTFSI-EC (ethylene carbonate) with the lithium metal anode. The simulation results show that TFSI anion prefers to be decomposed, while DME does not, thus, TFSI plays the vital role of protecting DME. However, in the LiTFSI-EC system, both TFSI anion and EC are decomposed, indicating that EC is less stable and not suitable to the formation of stable SEI. Thanks to the computational efficiency of the HAIRs method, we have completed the 1 ns simulation in a few days. Using the hardware, the above calculation would take at least one to two months if only the QM method was employed. Meanwhile the long HAIRs calculation shows that for the simulation of chemical reactions in SEI, at least 1 ns is essential. Instead, previous molecular dynamics (MD) simulations with a few ps, or tens of ps, are insufficient to fully capture the critical chemical reactions. The above simulation results provide reliable experience for the computational simulation study of SEI formation, and lay the theoretical foundation for the rational design of electrolytes and the development of high-performance electrolyte solution systems.
/
| 〈 |
|
〉 |
