
锐钛矿TiO2(101)表面电子能带结构的理论研究
收稿日期: 2016-04-20
修回日期: 2016-05-12
网络出版日期: 2016-05-16
基金资助
国家自然科学基金项目(No. 21373166)资助
Aligning Electronic Energy Levels on the Anatase TiO2(101) Surface
Received date: 2016-04-20
Revised date: 2016-05-12
Online published: 2016-05-16
赵俊杰 , 程俊 . 锐钛矿TiO2(101)表面电子能带结构的理论研究[J]. 电化学, 2017 , 23(1) : 45 -52 . DOI: 10.13208/j.electrochem.160418
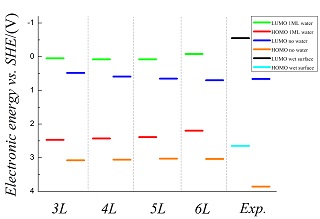
As one of the most commonly-used materials for photocatalysis and solar energy conversion, titanium dioxide (TiO2) has been extensively studied for more than 40 years. Its photoelectrochemical activity crucially depends on the band positions at the interface. In this work, the valence band maximum (VBM) and conduction band minimum (CBM) of a model TiO2 surface are computed using the standard work function method at the level of Perdew-Burke-Ernzerhof (PBE) density functional, which are then converted to the scale of the standard hydrogen electrode (SHE) by subtracting the absolute SHE potential. Comparing with the rutile TiO2(110) surface, we find a similar upshift in the VBM and CBM upon the adsorption of water molecules on the anatase TiO2(101) surface, and the band gap error intrinsic to the PBE functional can be mainly attributable to mis-positioning of the VBM. In addition, in contrast to the finding on the rutile TiO2(110) surface that the adsorption of 1 monolayer water largely recovers the band alignment of the aqueous interface, our preliminary calculations indicate that on the anatase TiO2(101) surface there is a considerable difference between the simplified model with the adsorption of 1 monolayer water and the fully solvated interface, suggesting the necessity to include the water molecules beyond the first adsorption layer in order to realistically represent the anatase TiO2 water interface.
/
| 〈 |
|
〉 |
