电化学(中英文) ›› 2021, Vol. 27 ›› Issue (1): 92-99. doi: 10.13208/j.electrochem.200621
蔡转运, 刘佳, 关思远, 吴德印*( ), 田中群
), 田中群
Zhuan-Yun Cai, Jia Liu, Si-Yuan Guan, De-Yin Wu*(), Zhong-Qun Tian
摘要:
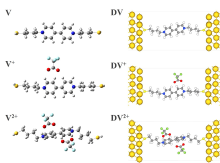
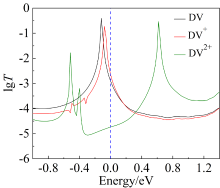
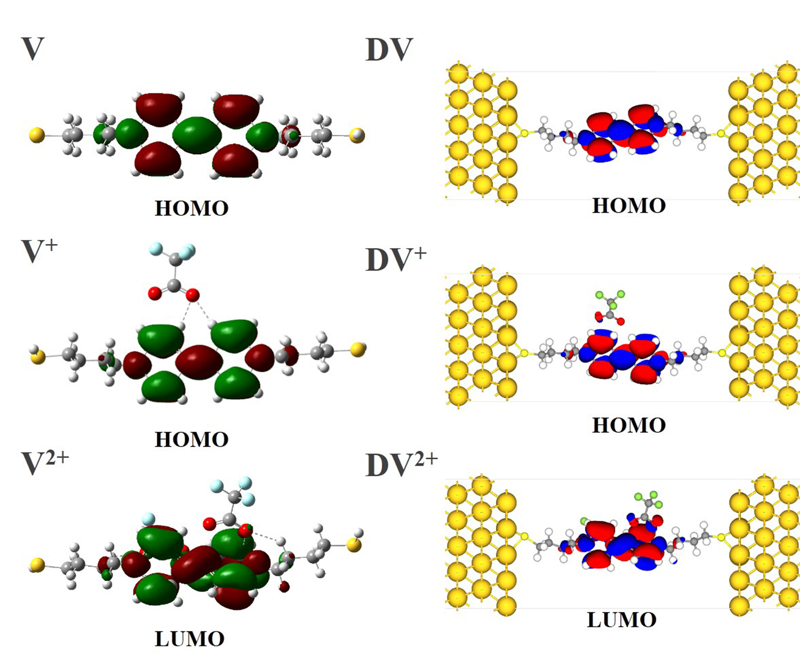
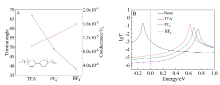
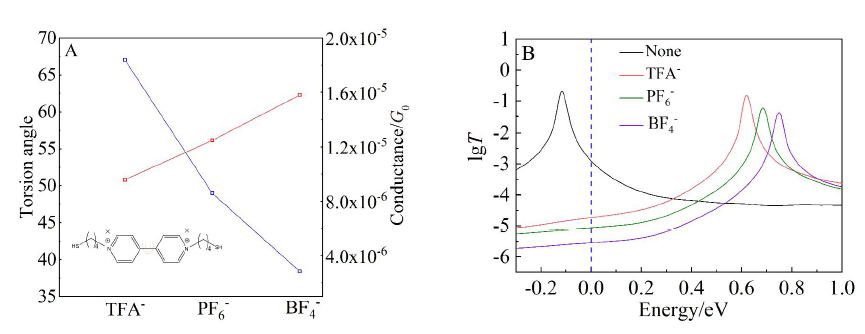
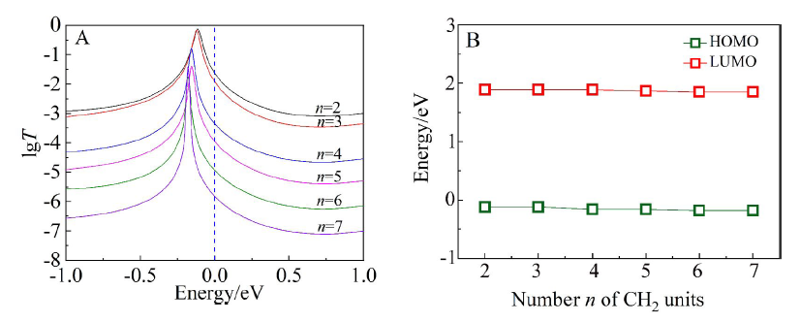
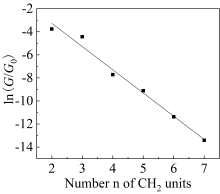
本文基于密度泛函(DFT)结合非平衡格林函数(NEGF)的方法,以具有氧化还原中心的紫罗碱衍生物(N,N′-bis(4-thioalkyl)-4,4′-bipyridinium, HS-4V4-SH)功能分子构造Au(111)/S-4V4-S/Au(111)分子结,详细分析了分子在三种价态V、V+和V2+下的电学性质与分子的几何结构和电子结构的关系。基于对三种价态透射系数分析结果表明,在零偏压下,V与V+的电导值比V2+高了两个数量级,4V4分子结的电导随两个吡啶环之间夹角的增大呈线性减小。同时,理论计算结果也表明,增加烷基链(HS-nVn-SH, n = 2 ~ 7)的数目,发现分子结电导值呈指数形式衰减,其每个亚甲基的衰减因子约为1,与烷基二硫醇分子的接近。