电化学(中英文) ›› 2024, Vol. 30 ›› Issue (2): 2307181. doi: 10.13208/j.electrochem.2307181
所属专题: “教程”专题文章
• 教程 • 上一篇
王锋a, 程俊a,b,c,*( )
)
收稿日期:2023-07-18
修回日期:2023-10-04
接受日期:2023-11-06
发布日期:2023-12-25
出版日期:2024-02-28
通讯作者:
*程俊,Tel: (86-592)2181570;E-mail: juncheng@xmu.edu.cn基金资助:
Feng Wanga, Jun Chenga,b,c,*()
Received:2023-07-18
Revised:2023-10-04
Accepted:2023-11-06
Online:2023-12-25
Published:2024-02-28
摘要:
氧化还原电位和酸度常数作为重要的物理化学性质被应用于分析能源材料重要指标值。为了实现能源材料的计算设计,发展计算电化学的方法,在复杂电化学环境下计算这些性质至关重要。近年来,利用计算电化学方法计算氧化还原电位和酸度常数已经受到了广泛的关注。然而,常用的计算方法如基于隐式溶剂化模型的小分子自由能计算,对于复杂溶剂化环境的处理非常有限。因此,基于第一性原理分子动力学(AIMD)的自由能计算被引入来描述复杂溶剂化环境中的溶质-溶剂相互作用。同时,基于AIMD的自由能计算方法已经被证实可以准确预测这些物理化学性质。然而,由于AIMD计算效率低且计算资源需求大,需要引入机器学习分子动力学(MLMD)加速计算。MLMD通过机器学习方法,构建模拟体系结构到第一性原理计算结果的一对一映射,可以在低成本下实现长时间尺度的AIMD。对于氧化还原电位和酸度常数计算,如何构建训练机器学习势函数模型所需的数据集至关重要。本文介绍了如何通过自动化工作流实现自由能计算势函数的自动化构建,通过机器学习分子动力学计算自由能并转化为对应的物理化学性质。
王锋, 程俊. 机器学习加速氧化还原电位和酸度常数计算[J]. 电化学(中英文), 2024, 30(2): 2307181.
Feng Wang, Jun Cheng. Automated Workflow for Redox Potentials and Acidity Constants Calculations from Machine Learning Molecular Dynamics[J]. Journal of Electrochemistry, 2024, 30(2): 2307181.
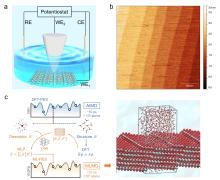
图1
机器学习势函数示意图[36]
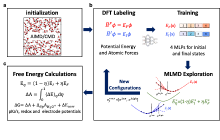
图2
自动化工作流的图示。(a) 初始化;(b) 势函数训练;(c)自由能计算[31]。
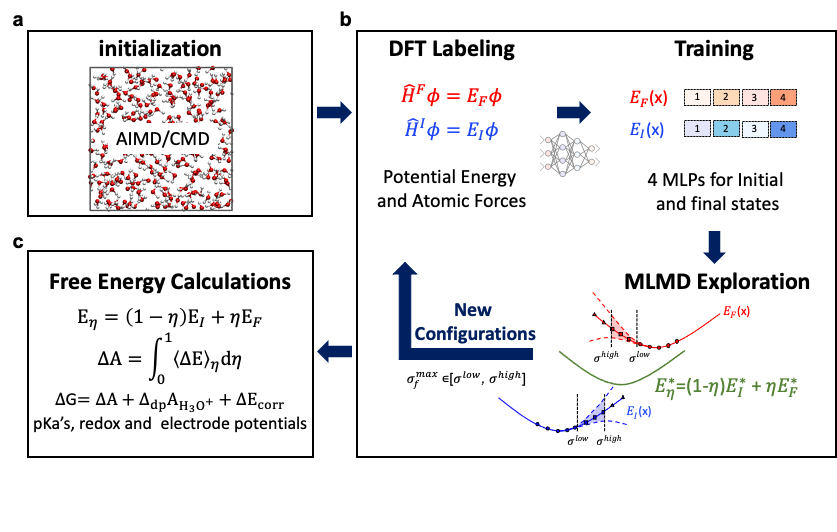
表1
通过自动化工作流获得的数据集大小和势函数对训练集的预测误差[31]
| 800, 800 | 0.553, 0.615 | 42.8, 44.2 | |
| 6016, 5234 | 0.653, 0.809 | 47.5, 50.8 | |
| 835, 835 | 0.580, 0.700 | 44.0, 45.7 | |
| 11048, 10648 | 0.556, 0.579 | 44.0, 43.4 | |
| 850, 827 | 0.544, 0.551 | 43.4, 45.5 | |
| 875, 873 | 0.532, 0.586 | 44.7, 48.1 | |
| 1996, 1996 | 0.797, 0.706 | 51.6, 49.5 | |
| 887, 887 | 0.722, 0.604 | 47.7, 47.8 | |
| 5499, 5499 | 0.819, 0.753 | 53.5, 52.1 | |
| 5791, 5791 | 0.733, 0.603 | 57.1, 47.7 |
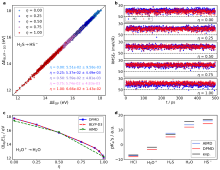
图3
(a) H2S → HS-体系,ΔdpE的准确性分析。(b) HCl → Cl-体系中每个 η预测原子受力的RMSE随模拟时间演变情况。(c) H3O+ → H2O体系AIMD、MLMD和基于MLMD轨迹的DFT计算得到的热力学积分曲线的对比。(d) 酸度常数的AIMD和MLMD的计算结果以及实验值。
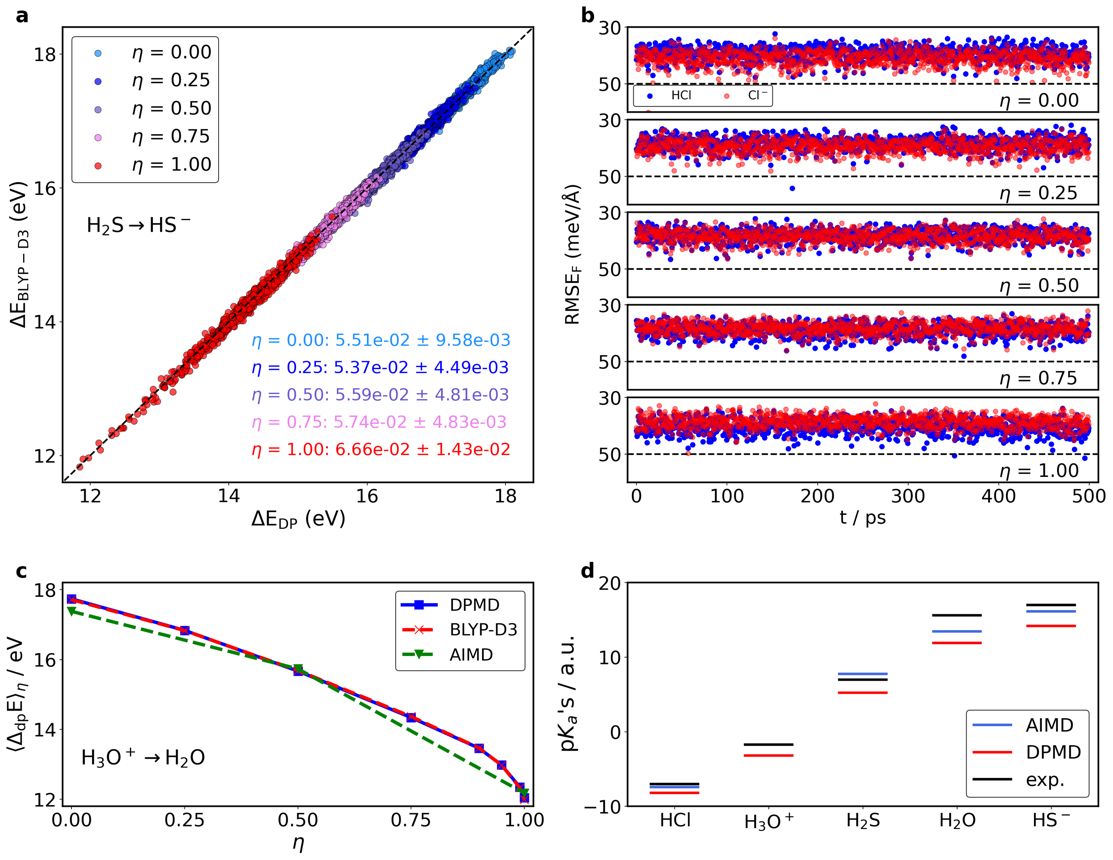
表2
通过基于MLMD模拟得到的酸度常数[31]
| AIMD | MLMD | MLMD* | exp. | |
|---|---|---|---|---|
| HCl | -7.0 | -8.2 (-8.8) | -6.7 (-7.3) | -7.0 |
| -3.2 | -3.2 (-3.2) | -1.7 (-1.7) | -1.7 | |
| 7.8 | 5.2 (5.0) | 6.7 (6.5) | 7.0 | |
| 13.5 | 11.9 (11.6) | 13.4 (13.1) | 15.6 | |
| 16.2 | 14.2 (14.5) | 15.7 (16.0) | 17.0 |
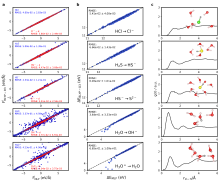
图4
不同体系脱质子自由能计算中η = 1.00的误差分析。(a) 原子受力误差分析。(b) 垂直能量差ΔEη=1.0的误差分析。(c) 虚原子和水分子上氢原子的径向分布函数[31]。
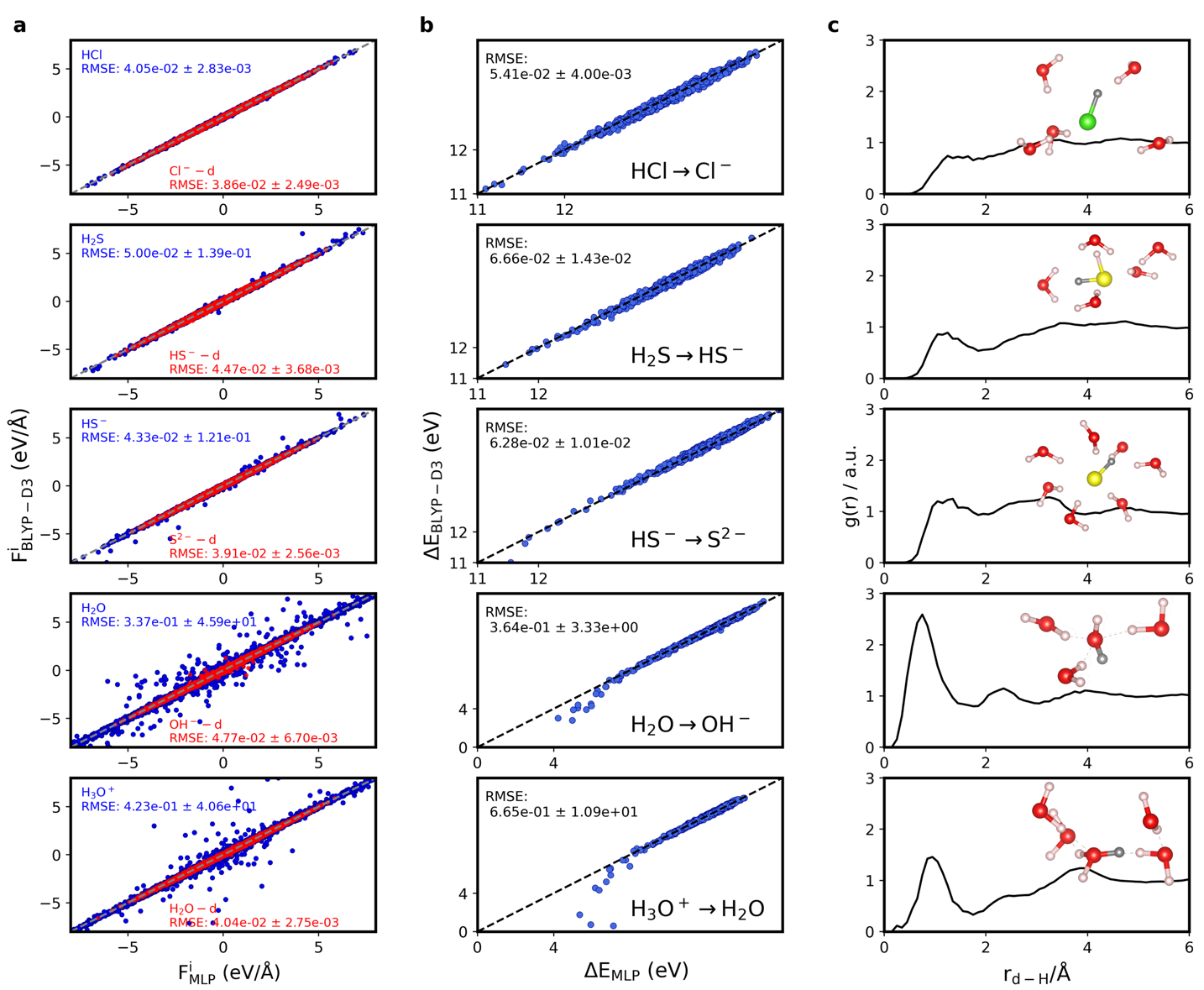
表3
通过基于MLMD模拟得到的氧化还原电位。MLMD计算使用的 Δ d p A H 3 O +为MLMD计算出的15.512 eV[31]。
| AIMD | MLMD (3P) | MLMD | exp. | |
|---|---|---|---|---|
| 1.5 | 1.58 | 1.84 | 2.41 | |
| 1.3 | 1.49 (1.51) | 1.68 (1.68) | 1.90 | |
| 0.5 | 0.69 | 0.76 | 1.08 | |
| -0.5 | -0.35 | -0.22 | -0.16 | |
| -2.07 | -1.98 | -2.06 | -1.90 |

图5
(a) 各个氧化还原自由能计算的热力学积分曲线。(b) MLMD计算得到的氧化还原电位。(c-f) O H ∙ / O H -热力学积分上η = 0.00, 0.75, 0.875和1.00上MLP预测的原子受力和DFT验证计算的对比[31]。
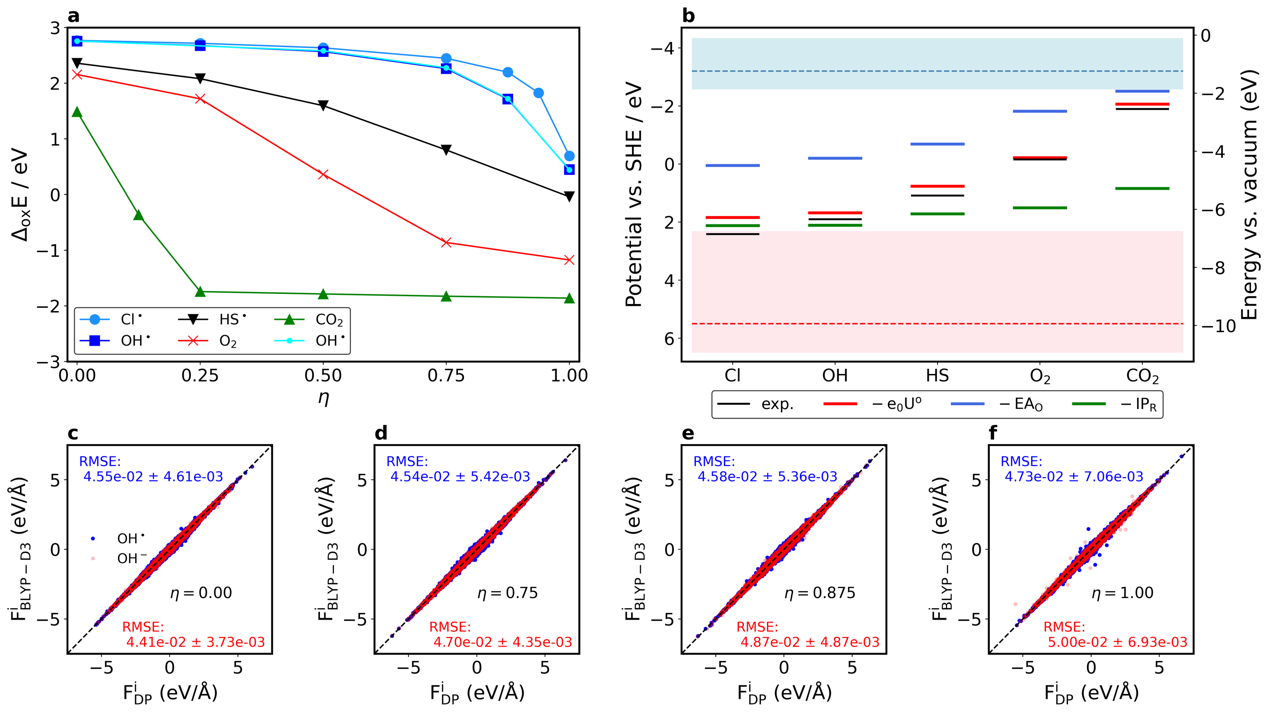
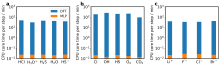
图6
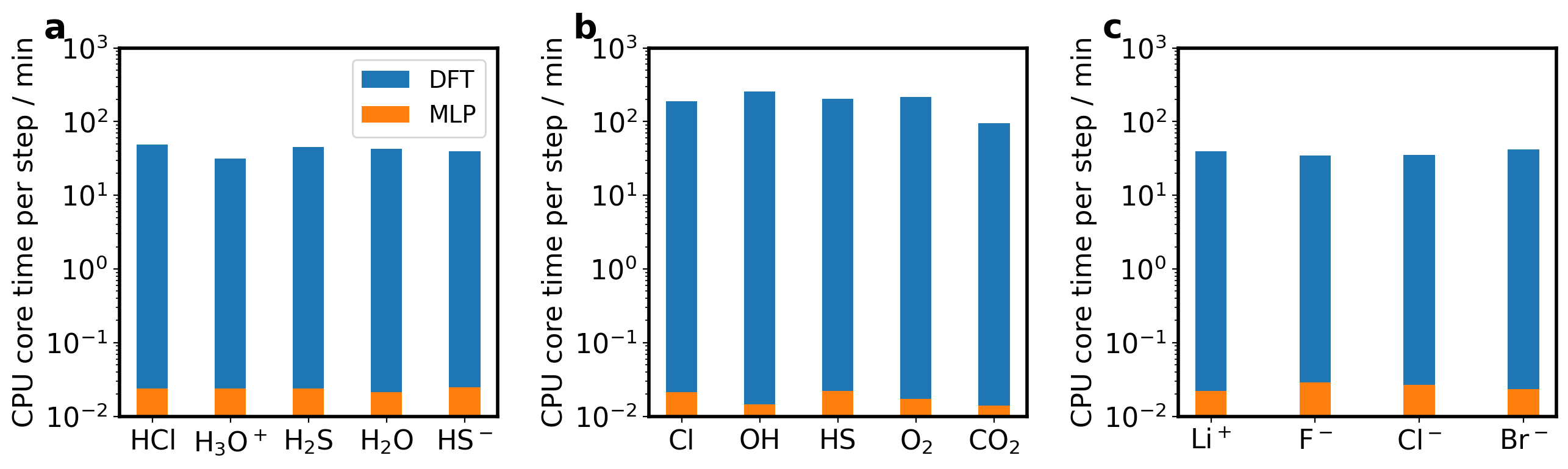
(a) 脱质子自由能计算和(b)氧化还原自由能计算中DFT和MLP计算效率对比。
| [1] |
Blumberger J. Recent advances in the theory and molecular simulation of biological electron transfer reactions[J]. Chem. Rev., 2015, 115(20): 11191-11238.
doi: 10.1021/acs.chemrev.5b00298 pmid: 26485093 |
| [2] |
Wardman P. Reduction potentials of one‐electron couples involving free radicals in aqueous solution[J]. J. Phys. Chem. Ref. Data, 1989, 18(4): 1637-1755.
doi: 10.1063/1.555843 URL |
| [3] |
Weinberg D R, Gagliardi C J, Hull J F, Murphy C F, Kent C A, Westlake B C, Meyer T J. Proton-coupled electron transfer[J]. Chem. Rev., 2012, 112(7): 4016-4093.
doi: 10.1021/cr200177j pmid: 22702235 |
| [4] |
Yu J, Zhao T S, Pan D. Tuning the performance of aqueous organic redox flow batteries via first-principles calculations[J]. J. Phys. Chem. Lett., 2020, 11(24): 10433-10438.
doi: 10.1021/acs.jpclett.0c03008 pmid: 33269931 |
| [5] |
Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models[J]. Chem. Rev., 2005, 105(8): 2999-3094.
doi: 10.1021/cr9904009 pmid: 16092826 |
| [6] |
Leung K. Predicting the voltage dependence of interfacial electrochemical processes at lithium-intercalated graphite edge planes[J]. Phys. Chem. Chem. Phys., 2015, 17(3): 1637-1643.
doi: 10.1039/c4cp04494k pmid: 25438093 |
| [7] |
Le J, Iannuzzi M, Cuesta A, Cheng J. Determining potentials of zero charge of metal electrodes versus the standard hydrogen electrode from density-functional-theory-based molecular dynamics[J]. Phys. Rev. Lett., 2017, 119(1): 016801.
doi: 10.1103/PhysRevLett.119.016801 URL |
| [8] |
Rossmeisl J, Skúlason E, Björketun M E, Tripkovic V, Nørskov J K. Modeling the electrified solid-liquid interface[J]. Chem. Phys. Lett., 2008, 466(1-3): 68-71.
doi: 10.1016/j.cplett.2008.10.024 URL |
| [9] |
Zhang C, Sprik M. Finite field methods for the supercell modeling of charged insulator/electrolyte interfaces[J]. Phys. Rev. B, 2016, 94(24): 245309.
doi: 10.1103/PhysRevB.94.245309 URL |
| [10] |
King G, Warshel A. Investigation of the free energy functions for electron transfer reactions[J]. J. Chem. Phys., 1990, 93(12): 8682-8692.
doi: 10.1063/1.459255 URL |
| [11] |
Cheng J, Sulpizi M, Sprik M. Redox potentials and pKa for benzoquinone from density functional theory based molecular dynamics[J]. J. Chem. Phys., 2009, 131(15): 154504.
doi: 10.1063/1.3250438 URL |
| [12] |
Blumberger J, Tavernelli I, Klein M L, et al. Diabatic free energy curves and coordination fluctuations for the aqueous Ag+/Ag2+ redox couple: A biased born-oppenheimer molecular dynamics investigation[J]. J Chem. Phys., 2006, 124(6): 064507.
doi: 10.1063/1.2162881 URL |
| [13] | Costanzo F, Sulpizi M, Valle R G D, Sprik M. The oxidation of tyrosine and tryptophan studied by a molecular dynamics normal hydrogen electrode[J]. J. Chem. Phys., 2011, 134(24): 06B615. |
| [14] |
Cheng J, Liu X, VandeVondele J, Sulpizi M, Sprik M. Redox potentials and acidity constants from density functional theory based molecular dynamics[J]. Acc. Chem. Res., 2014, 47(12): 3522-3529.
doi: 10.1021/ar500268y URL |
| [15] |
Yang X H, Cuesta A, Cheng J. Computational Ag/AgCl reference electrode from density functional theory-based molecular dynamics[J]. J. Phys. Chem. B, 2019, 123(48): 10224-10232.
doi: 10.1021/acs.jpcb.9b06650 URL |
| [16] |
Leung K, Tenney C M. Toward first principles prediction of voltage dependences of electrolyte/electrolyte interfacial processes in lithium ion batteries[J]. J. Phys. Chem. C, 2013, 117(46): 24224-24235.
doi: 10.1021/jp408974k URL |
| [17] |
Cheng J, VandeVondele J. Calculation of electrochemical energy levels in water using the random phase approximation and a double hybrid functional[J]. Phys. Rev. Lett., 2016, 116(8): 086402.
doi: 10.1103/PhysRevLett.116.086402 URL |
| [18] | Su N Q, Xu X. The XYG3 type of doubly hybrid density functionals[J]. WIRES. Co. Mol. Sci., 2016, 6(6): 721-747. |
| [19] |
Su N Q, Zhu Z, Xu X. Doubly hybrid density functionals that correctly describe both density and energy for atoms[J]. P. Natl. Acad. Sci., 2018, 115(10): 2287-2292.
doi: 10.1073/pnas.1713047115 URL |
| [20] |
Zhang I Y, Wu J, Xu X. Accurate heats of formation of polycyclic saturated hydrocarbons predicted by using the XYG3 type of doubly hybrid functionals[J]. J. Comput. Chem., 2019, 40(10): 1113-1122.
doi: 10.1002/jcc.25726 pmid: 30379331 |
| [21] | Thompson A P, Swiler L P, Trott C R, Foiles S M, Tucker G J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials[J]. J. Comput. Phys., 2015, 285: 316-330. |
| [22] |
Huan T D, Batra R, Chapman J, Krishnan S, Chen L, Ramprasad R. A universal strategy for the creation of machine learning-based atomistic force fields[J]. NPJ Comput. Mater., 2017, 3(1): 1-8.
doi: 10.1038/s41524-016-0004-9 |
| [23] |
Behler J, Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces[J]. Phys. Rev. Lett., 2007, 98(14): 146401.
doi: 10.1103/PhysRevLett.98.146401 URL |
| [24] |
Behler J. Perspective: Machine learning potentials for atomistic simulations[J]. J. Chem. Phys., 2016, 145(17): 170901.
doi: 10.1063/1.4966192 URL |
| [25] |
Bartók A P, Payne M C, Kondor R, Csányi G. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons[J]. Phys. Rev. Lett., 2010, 104(13): 136403.
doi: 10.1103/PhysRevLett.104.136403 URL |
| [26] |
Rupp M, Tkatchenko A, Müller K R, Von Lilienfeld O A. Fast and accurate modeling of molecular atomization energies with machine learning[J]. Phys. Rev. Lett., 2012, 108(5): 058301.
doi: 10.1103/PhysRevLett.108.058301 URL |
| [27] |
Zhang L, Han J, Wang H, Car R, Weinan E. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics[J]. Phys. Rev. Lett., 2018, 120(14): 143001.
doi: 10.1103/PhysRevLett.120.143001 URL |
| [28] |
Wang H, Zhang L, Han J, Weinan E. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics[J]. Comput. Phys. Commun., 2018, 228: 178-184.
doi: 10.1016/j.cpc.2018.03.016 URL |
| [29] |
Chmiela S, Tkatchenko A, Sauceda H E, Poltavsky I, Schütt K T, Müller K R. Machine learning of accurate energy-conserving molecular force fields[J]. Sci. Adv., 2017, 3(5): e1603015.
doi: 10.1126/sciadv.1603015 URL |
| [30] |
Schütt K T, Arbabzadah F, Chmiela S, Müller K R, Tkatchenko A. Quantum-chemical insights from deep tensor neural networks[J]. Nat. Commun., 2017, 8(1): 1-8.
doi: 10.1038/s41467-016-0009-6 |
| [31] |
Wang F, Cheng J. Automated workflow for computation of redox potentials, acidity constants, and solvation free energies accelerated by machine learning[J]. J. Chem. Phys. 2022; 157(2): 024103
doi: 10.1063/5.0098330 URL |
| [32] | Zhang L, Lin D Y, Wang H, Car R, Weinan E. Active learning of uniformly accurate interatomic potentials for materials simulation[J]. Phys. Rev. Mater., 2019, 3(2): 023804. |
| [33] |
Zhang Y, Wang H, Chen W, Zeng J, Zhang L, Wang H, Weinan E. DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models[J]. Comput. Phys. Commun., 2020, 253: 107206.
doi: 10.1016/j.cpc.2020.107206 URL |
| [34] |
Kirkwood J G. Statistical mechanics of fluid mixtures[J]. J. Chem. Phys., 1935, 3(5): 300-313.
doi: 10.1063/1.1749657 URL |
| [35] |
Sulpizi M, Sprik M. Acidity constants from vertical energy gaps: density functional theory based molecular dynamics implementation[J]. Phys. Chem. Chem. Phys., 2008, 10(34): 5238-5249.
doi: 10.1039/b802376j pmid: 18728866 |
| [36] | Sun Y, Wu C R, Wang F, Bi R H, Zhuang Y B, Liu S, Cheng J. Step-induced double-row pattern of interfacial water on rutile TiO2 (110) at electrochemical conditions[J]. doi: 10.26434/chemrxiv-2023-7wsqv 2023. |
| [37] |
VandeVondele J, Krack M, Mohamed F, Parrinello M, Chassaing T, Hutter J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach[J]. Comput. Phys. Commun., 2005, 167(2): 103-128.
doi: 10.1016/j.cpc.2004.12.014 URL |
| [38] |
Becke A D. Density-functional exchange-energy approximation with correct asymptotic behavior[J]. Phys. Rev. A, 1988, 38(6): 3098.
doi: 10.1103/PhysRevA.38.3098 URL |
| [39] |
Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J]. Phys. Rev. B, 1988, 37(2): 785.
doi: 10.1103/physrevb.37.785 pmid: 9944570 |
| [40] |
Grimme S, Antony J, Ehrlich S & Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu[J]. J. Chem. Phys., 2010, 132(15): 154104.
doi: 10.1063/1.3382344 URL |
| [41] |
Hartwigsen C, Gœdecker S, Hutter J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn[J]. Phys. Rev. B, 1998, 58(7): 3641.
doi: 10.1103/PhysRevB.58.3641 URL |
| [42] |
Goedecker S, Teter M, Hutter J. Separable dual-space Gaussian pseudopotentials[J]. Phys. Rev. B, 1996, 54(3): 1703.
pmid: 9986014 |
| [43] |
VandeVondele J, Hutter J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases[J]. J. Chem. Phys., 2007, 127(11): 114105.
doi: 10.1063/1.2770708 URL |
| [44] | Zhang L, Han J, Wang H, Saidi W & Car R. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems[J]. Adv. Neural Inf. Process. Syst., 2018: 31. |
| [45] |
Sulpizi M, Sprik M. Acidity constants from vertical energy gaps: density functional theory based molecular dynamics implementation[J]. Phys. Chem. Chem. Phys., 2008, 10(34): 5238-5249.
doi: 10.1039/b802376j pmid: 18728866 |
| [1] | 姜晶, 王佳, . 液相分散程度对气/液/固多相体系腐蚀行为的影响[J]. 电化学(中英文), 2009, 15(2): 135-140. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||

