
电化学(中英文) ›› 2021, Vol. 27 ›› Issue (1): 92-99. doi: 10.13208/j.electrochem.200621
所属专题: “理论计算模拟”专题文章
蔡转运, 刘佳, 关思远, 吴德印*( ), 田中群
), 田中群
收稿日期:2020-06-21
修回日期:2020-07-15
出版日期:2021-02-28
发布日期:2020-07-16
通讯作者:
吴德印
E-mail:dywu@xmu.edu.cn
基金资助:
Zhuan-Yun Cai, Jia Liu, Si-Yuan Guan, De-Yin Wu*(), Zhong-Qun Tian
Received:2020-06-21
Revised:2020-07-15
Published:2021-02-28
Online:2020-07-16
Contact:
De-Yin Wu
E-mail:dywu@xmu.edu.cn
摘要:
本文基于密度泛函(DFT)结合非平衡格林函数(NEGF)的方法,以具有氧化还原中心的紫罗碱衍生物(N,N′-bis(4-thioalkyl)-4,4′-bipyridinium, HS-4V4-SH)功能分子构造Au(111)/S-4V4-S/Au(111)分子结,详细分析了分子在三种价态V、V+和V2+下的电学性质与分子的几何结构和电子结构的关系。基于对三种价态透射系数分析结果表明,在零偏压下,V与V+的电导值比V2+高了两个数量级,4V4分子结的电导随两个吡啶环之间夹角的增大呈线性减小。同时,理论计算结果也表明,增加烷基链(HS-nVn-SH, n = 2 ~ 7)的数目,发现分子结电导值呈指数形式衰减,其每个亚甲基的衰减因子约为1,与烷基二硫醇分子的接近。
蔡转运, 刘佳, 关思远, 吴德印, 田中群. 紫罗碱衍生物分子结的电学性质理论研究[J]. 电化学(中英文), 2021, 27(1): 92-99.
Zhuan-Yun Cai, Jia Liu, Si-Yuan Guan, De-Yin Wu, Zhong-Qun Tian. Theoretical Study on Electrical Properties of Molecular Junctions of Viologen Derivatives[J]. Journal of Electrochemistry, 2021, 27(1): 92-99.
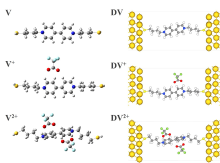
图1
HS-4V4-SH三种价态的分子与其对应的分子结结构(网络版彩图)

表1
六种结构中吡啶环之间碳碳键长、二面角、Mulliken电荷与NBO电荷
| State | C-C bond (Å) | Dihedra angle | Mulliken charge (elec.) | NBO charge (elec.) |
|---|---|---|---|---|
| V | 1.382 | 0.2° | - | - |
| V+ | 1.431 | 0.9° | 0.892 | 0.936 |
| V2+ | 1.479 | 51.0° | 1.705 | 1.884 |
| DV | 1.391 | 0.3° | - | - |
| DV+ | 1.446 | 1.3° | 0.823 | - |
| DV2+ | 1.479 | 50.8° | 1.670 | - |
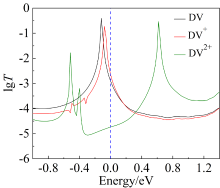
图2
三种价态分子结的透射谱图(网络版彩图)


图3
分子与分子结中分子的HOMO与LUMO能级电子结构。(网络版彩图)
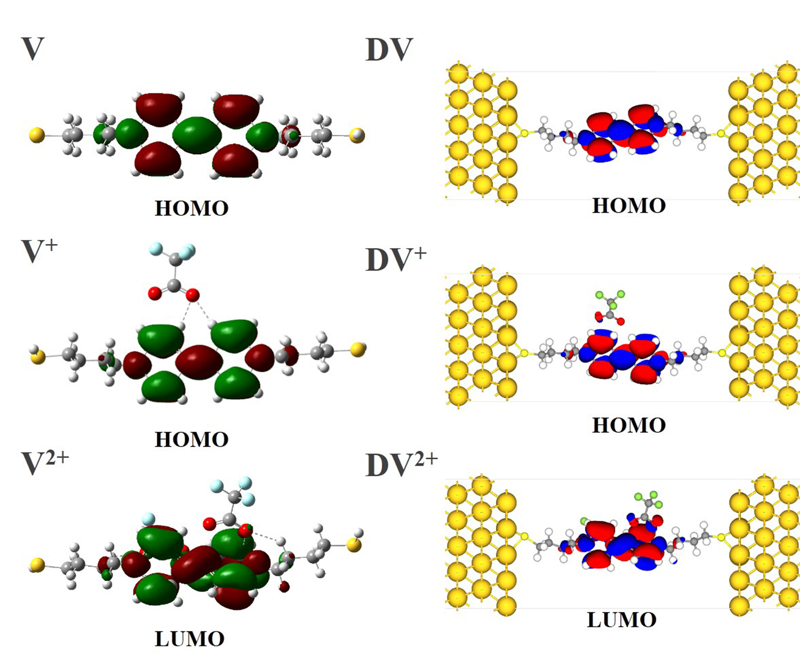
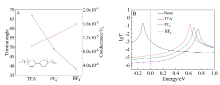
图4
A 引入不同离子对分子电导和吡啶环之间扭转角的影响;B. 分子结的透射谱图。(网络版彩图)
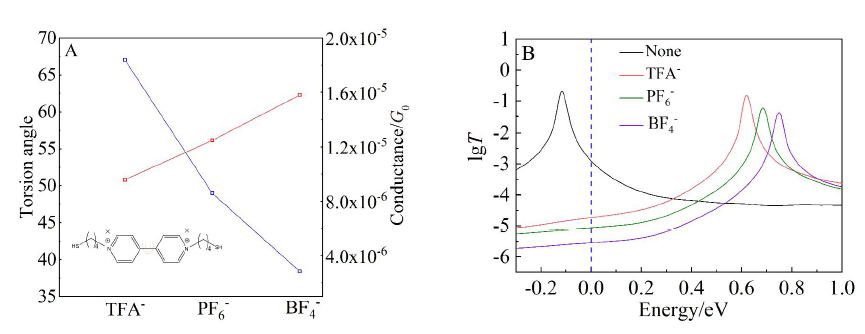

图5
A. 不同碳链长的透射谱图;B. 分子前线轨道能级随烷基链长的变化(网络版彩图)
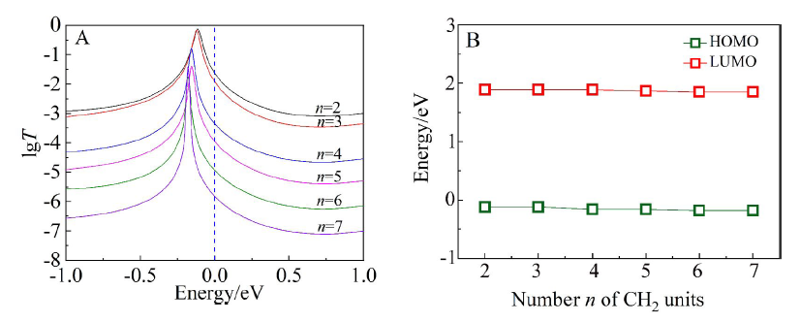
表2
不同烷基链分子长度(两端S原子之间的长度)零偏压电导值(G0 ≈ 77.4 μS)
| n | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| L/nm | 1.493 | 1.600 | 1.977 | 2.098 | 2.437 | 2.631 |
| G/G0 | 2.31×10-2 | 1.19×10-2 | 4.44×10-4 | 1.10×10-4 | 1.15×10-5 | 1.50×10-6 |
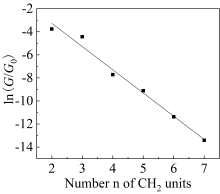
图6
分子电导值与CH2数目关系的指数拟合曲线

| [1] | Moore G E. Cramming more components onto integrated circuits[J]. Electronics, 1965,38(8):114-117. |
| [2] |
Jia C C, Ma B J, Xin N, Guo X F. Carbon electrode-molecule junctions: A reliable platform for molecular electronics[J]. Acc. Chem. Res., 2015,48(9):2565-2575.
doi: 10.1021/acs.accounts.5b00133 URL pmid: 26190024 |
| [3] |
Aviram A, Ratner M A. Molecular rectifiers[J]. Chem. Phys. Lett., 1974,29(2):277-283.
doi: 10.1016/0009-2614(74)85031-1 URL |
| [4] |
Xin N, Guan J X, Zhou C G, et al. Concepts in the design and engineering of single-molecule electronic devices[J]. Nat. Rev. Phys., 2019,1(3):211-230.
doi: 10.1038/s42254-019-0022-x URL |
| [5] |
Jia C C, Migliore A, Xin N, Huang S Y, Wang J Y, Yan Q, Wang S P, Chen H L, Wang D M, Feng B Y, Liu Z R, Zhang G Y, Qu D H, Tian H, Ratner M A, Xu H Q, Nitzan A, Guo X F. Covalently bonded single-molecule junctions with stable and reversible photoswitched conductivity[J]. Science, 2016,352(6292):1443-1445.
doi: 10.1126/science.aaf6298 URL pmid: 27313042 |
| [6] |
Tan Z B, Zhang D, Tian H R, Wu Q Q, Hou S J, Pi J C, Sadeghi H, Tang Z, Yang Y, Liu J Y, Tan Y Z, Chen Z B, Shi J, Xiao Z Y, Lambert C, Xie S Y, Hong W J. Atomically defined angstrom-scale all-carbon junctions[J]. Nat. Commun., 2019,10(1):1748.
doi: 10.1038/s41467-019-09793-8 URL pmid: 30988310 |
| [7] |
Yang Y(杨扬), Liu J Y(刘俊扬), Yan R W(晏润文), Wu D Y(吴德印), Tian Z Q(田中群). Mechanism and characterization of electron transport through metal/molecule/metal junctions[J]. Chem. J. Chin. Univ.-Chin. (高等学校化学学报), 2015,36(1):9-23.
doi: 10.7503/cjcu20140941 URL |
| [8] |
Qi Y H, Guan D R, Liu C B. DFT study of the transport properties of molecular wire at low bias[J]. Chin. J. Chem., 2006,24(3):326-330.
doi: 10.1002/(ISSN)1614-7065 URL |
| [9] | He Y Y(贺园园), Zhao J W(赵健伟). Effects of conformational transformations on electronic transport properties of optical molecular switches: An ab initio study[J]. J. Electrochem. (电化学), 2014,20(3):243-259. |
| [10] |
Zhou C, Li X X, Gong Z L, Jia C C, Lin Y W, Gu C H, He G, Zhong Y W, Yang J L, Guo X F. Direct observation of single-molecule hydrogen-bond dynamics with single-bond resolution[J]. Nat. Commun., 2018,9(1):807.
doi: 10.1038/s41467-018-03203-1 URL pmid: 29476061 |
| [11] |
Jöckel F, Watson M D, Müllen K, Rabe J P. Prototypical single-molecule chemical-field-effect transistor with nanometer-sized gates[J]. Phys. Rev. Lett., 2004,92(18):188303.
doi: 10.1103/PhysRevLett.92.188303 URL pmid: 15169538 |
| [12] |
Liu B, Blaszczyk A, Mayor M, Wandlowski T. Redox-switching in a viologen-type adlayer: An electrochemical shell-isolated nanoparticle enhanced raman spectroscopy study on Au(111)-(1×1) single crystal electrodes[J]. ACS Nano, 2011,5(7):5662-5672.
doi: 10.1021/nn201307g URL pmid: 21634391 |
| [13] |
Li J H, Cheng G J, Dong S J. Electrochemical study of the interfacial characteristics of redox-active viologen thiol self-assembled monolayers[J]. Thin Solid Films, 1997,293(1):200-205.
doi: 10.1016/S0040-6090(96)08995-X URL |
| [14] |
Osorio H M, Martín S, Milan D C, Gonzalez-Orive A, Gluyas JBG, Higgins S J, Low P J, Nichols R J, Cea P. Influence of surface coverage on the formation of 4,4′-bipyridinium (viologen) single molecular junctions[J]. J. Mater. Chem. C, 2017,5(45):11717-11723.
doi: 10.1039/C7TC03624H URL |
| [15] | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, revision D. 01[M]. Wallingford, CT; Gaussian, Inc., Wallingford CT, 2009. |
| [16] |
Becke A D. Density-functional thermochemistry. III. The role of exact exchange[J]. J. Chem. Phys., 1993,98(7):5648-5652.
doi: 10.1063/1.464913 URL |
| [17] | Krishnan R, Binkley J S, Seeger R, Pople J A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions[J]. J. Chem. Phys., 1980,72(1):650-654. |
| [18] |
McLean A D, Chandler G S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18[J]. J. Chem. Phys., 1980,72(10):5639-5648.
doi: 10.1063/1.438980 URL |
| [19] |
Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Phys. Rev., 1965,140(4A):A1133-A1138.
doi: 10.1103/PhysRev.140.A1133 URL |
| [20] |
Calais J L. Density-functional theory of atoms and mole-cules[J]. Int. J. Quantum Chem., 1993,47(1):101-101.
doi: 10.1002/qua.560470107 URL |
| [21] |
Becke A D. Perspective: Fifty years of density-functional theory in chemical physics[J]. J. Chem. Phys. , 2014,140(18):18A301.
URL pmid: 24832308 |
| [22] |
Thygesen K S. Electron transport through an interacting region: The case of a nonorthogonal basis set[J]. Phys. Rev. B , 2006,73(3):035309.
doi: 10.1103/PhysRevB.73.035309 URL |
| [23] |
Gruss D, Velizhanin K A, Zwolak M. Landauer's formula with finite-time relaxation: Kramers' crossover in electronic transport[J]. Sci. Rep., 2016,6:24514-24514.
doi: 10.1038/srep24514 URL pmid: 27094206 |
| [24] |
Ernzerhof M, Perdew J P. Generalized gradient approximation to the angle- and system-averaged exchange hole[J]. J. Chem. Phys., 1998,109(9):3313-3320.
doi: 10.1063/1.476928 URL |
| [25] |
Perdew J P, Chevary J A, Vosko S H, Jackson K A, Pederson M R, Singh D J, Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation[J]. Phys. Rev. B, 1992,46(11):6671-6687.
doi: 10.1103/PhysRevB.46.6671 URL |
| [26] |
Becke A D. Density functional calculations of molecular bond energies[J]. J. Chem. Phys., 1986,84(8):4524-4529.
doi: 10.1063/1.450025 URL |
| [27] |
Becke A D. Density-functional exchange-energy approximation with correct asymptotic behavior[J]. Phys. Rev. A, 1988,38(6):3098-3100.
doi: 10.1103/PhysRevA.38.3098 URL |
| [28] |
Hoft R, Ford M, García-Suárez V, Lambert C J. The effect of stretching thiyl- and ethynyl-Au molecular junctions[J]. J. Phys.-Condes. Matter , 2007,20(2):025207.
doi: 10.1088/0953-8984/20/02/025207 URL |
| [29] |
Taylor J, Guo H, Wang J. Ab initio modeling of quantum transport properties of molecular electronic devices[J]. Phys. Rev. B, 2001,63(24):245407.
doi: 10.1103/PhysRevB.63.245407 URL |
| [30] |
Meir Y, Wingreen N S. Landauer formula for the current through an interacting electron region[J]. Phys. Rev. Lett., 1992,68(16):2512-2515.
URL pmid: 10045416 |
| [31] |
Kim Y H, Tahir-Kheli J, Schultz P A, Goddard W A. First-principles approach to the charge-transport characteristics of monolayer molecular-electronics devices: Application to hexanedithiolate devices[J]. Phys. Rev. B, 2006,73(23):235419.
doi: 10.1103/PhysRevB.73.235419 URL |
| [32] |
Landauer R, Martin T. Barrier interaction time in tunneling[J]. Rev. Mod. Phys., 1994,66(1):217-228.
doi: 10.1103/RevModPhys.66.217 URL |
| [33] | Datta S. Quantum transport: atom to transistor[M]. Cambridge university press, 2005. |
| [34] |
Haiss W, van Zalinge H, Higgins S J, Bethell D, Hobenreich H, Schiffrin D J, Nichols R J. Redox state dependence of single molecule conductivity[J]. J. Am. Chem. Soc., 2003,125(50):15294-15295.
doi: 10.1021/ja038214e URL pmid: 14664565 |
| [35] |
Magoga M, Joachim C. Conductance and transparence of long molecular wires[J]. Phys. Rev. B, 1997,56(8):4722-4729.
doi: 10.1103/PhysRevB.56.4722 URL |
| [36] |
Samanta M P, Tian W, Datta S, Kubiak C P. Electronic conduction through organic molecules[J]. Phys. Rev. B, 1996,53(12):R7626-R7629.
doi: 10.1103/PhysRevB.53.R7626 URL |
| [37] |
Li Z, Pobelov I, Han B, Wandlowski T, Blaszczyk A, Mayor M. Conductance of redox-active single molecular junctions: an electrochemical approach[J]. Nanotechnology, 2006,18(4):044018.
doi: 10.1088/0957-4484/18/4/044018 URL |
| [38] |
Li C, Pobelov I, Wandlowski T, Bagrets A, Arnold A, Evers F. Charge transport in single Au | alkanedithiol | Au junctions: coordination geometries and conformational degrees of freedom[J]. J. Am. Chem. Soc., 2008,130(1):318-326.
doi: 10.1021/ja0762386 URL pmid: 18076172 |
| [39] |
Xu B, Tao N J. Measurement of single-molecule resistance by repeated formation of molecular junctions[J]. Science, 2003,301(5637):1221.
doi: 10.1126/science.1087481 URL pmid: 12947193 |
| [40] |
Li X L, He J, Hihath J, Xu B Q, Lindsay S M, Tao N J. Conductance of single alkanedithiols: Conduction mechanism and effect of molecule-electrode contacts[J]. J. Am. Chem. Soc., 2006,128(6):2135-2141.
doi: 10.1021/ja057316x URL pmid: 16464116 |
| [41] |
Guo S, Hihath J, Díez-Pérez I, Tao N J. Measurement and statistical analysis of single-molecule current-voltage characteristics, transition voltage spectroscopy, and tunneling barrier height[J]. J. Am. Chem. Soc., 2011,133(47):19189-19197.
URL pmid: 21991939 |
| [42] |
Yan R W, Jin X, Guan S Y, Zhang X G, Pang R, Tian Z Q, Wu D Y, Mao B W. Theoretical study of quantum conductance of conjugated and nonconjugated molecular wire junctions[J]. J. Phys. Chem. C, 2016,120(22):11820-11830.
doi: 10.1021/acs.jpcc.6b03116 URL |
| No related articles found! |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||