
固态锂电池中正极/电解质界面的密度泛函计算研究
收稿日期: 2017-01-10
修回日期: 2017-02-22
网络出版日期: 2017-02-24
基金资助
This work was supported by the National Natural Science Foundation of China (Grants No. 11234013), “863” Project (Grant No. 2015AA034201), Beijing S&T Project (Grant No. D161100002416003), and Youth Innovation Promotion Association (Grant No.2016005) for financial support and the Shanghai Supercomputer Center for providing computing resources.
Density functional investigation on cathode/electrolyte interface in solid-state lithium batteries
Received date: 2017-01-10
Revised date: 2017-02-22
Online published: 2017-02-24
Supported by
This work was supported by the National Natural Science Foundation of China (Grants No. 11234013), “863” Project (Grant No. 2015AA034201), Beijing S&T Project (Grant No. D161100002416003), and Youth Innovation Promotion Association (Grant No.2016005) for financial support and the Shanghai Supercomputer Center for providing computing resources.
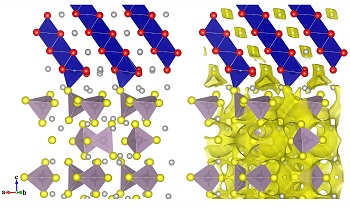
锂离子电池的广泛应用对储能器件的能量密度、安全性和充放电速度提出了新的要求. 全固态锂电池与传统锂离子电池相比具有更少的副反应和更高的安全性,已成为下一代储能器件的首选. 构建匹配的电极/电解质界面是在全固态锂电池中获得优异综合性能的关键. 本文采用第一性原理计算研究了固态电池中电解质表面及正极/电解质界面的局域结构和锂离子输运性质. 选取β-Li3PS4 (010)/LiCoO2 (104)和 Li4GeS4 (010)/LiCoO2 (104)体系计算了界面处的成键情况及锂离子的迁移势垒. 部分脱锂态的正极/电解质界面上由于Co-S成键的加强削弱了P/Ge-S键的强度,降低了对Li+的束缚,从而导致了更低的锂离子迁移势垒. 理解界面局域结构及其对Li+输运性质的影响将有助于我们在固态电池中构建性能优异的电极/电解质界面.
王雪龙, 肖睿娟, 向勇, 李泓, 陈立泉 . 固态锂电池中正极/电解质界面的密度泛函计算研究[J]. 电化学, 2017 , 23(4) : 381 -390 . DOI: 10.13208/j.electrochem.170142
The rapidly expanding application of lithium ion batteries stimulates research interest on energy storage devices with higher energy density, better safety and faster charge/discharge speed. All-solid-state lithium batteries have been considered as promising candidates because of their fewer side reactions and better safety compared with conventional lithium-ion batteries with organic liquid electrolytes. Looking for well-matched electrode/electrolyte interfaces is one of the keys to ensuring good comprehensive performance of solid-state lithium batteries. In this report, with the aid of first-principles simulations, the local structure and lithium ions transportation properties of electrolyte surfaces and cathode/electrolyte interfaces are investigated. The β-Li3PS4 (010)/LiCoO2 (104) and Li4GeS4(010)/LiCoO2(104) interfaces are adopted as model systems to understand the bonding interaction and Li+ migration barriers at interfaces. The ability of Li+ motion is improved in partial delithiated state for both systems, due to that Co atoms at the interface in high oxidized state oxidize the S atoms nearby and weaken the P/Ge-S bond resulting in less constrains on Li ions in neighbor and promoting the exchange of Li ions across the interface. It provides information for cathode/electrolyte interface optimization, and may help us discover appropriate techniques for solid-state lithium batteries.
/
| 〈 |
|
〉 |
