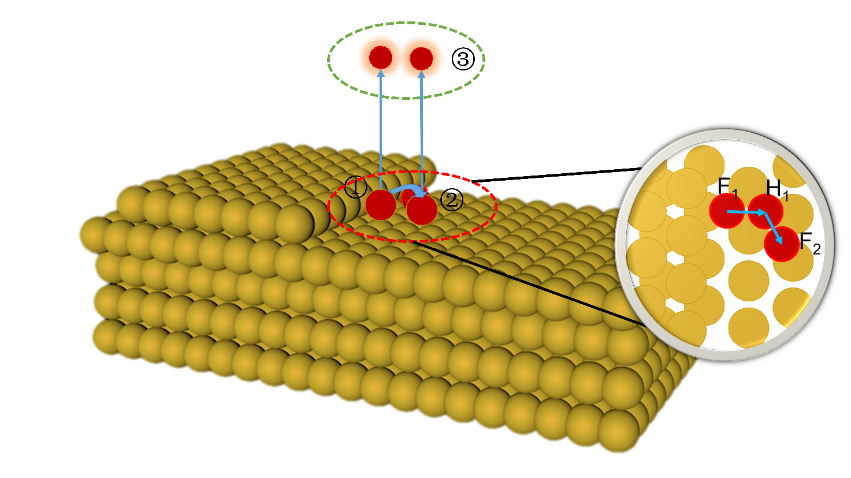
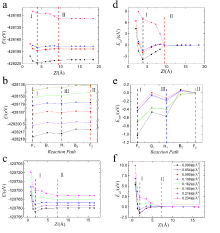
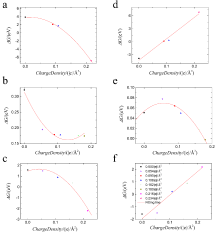
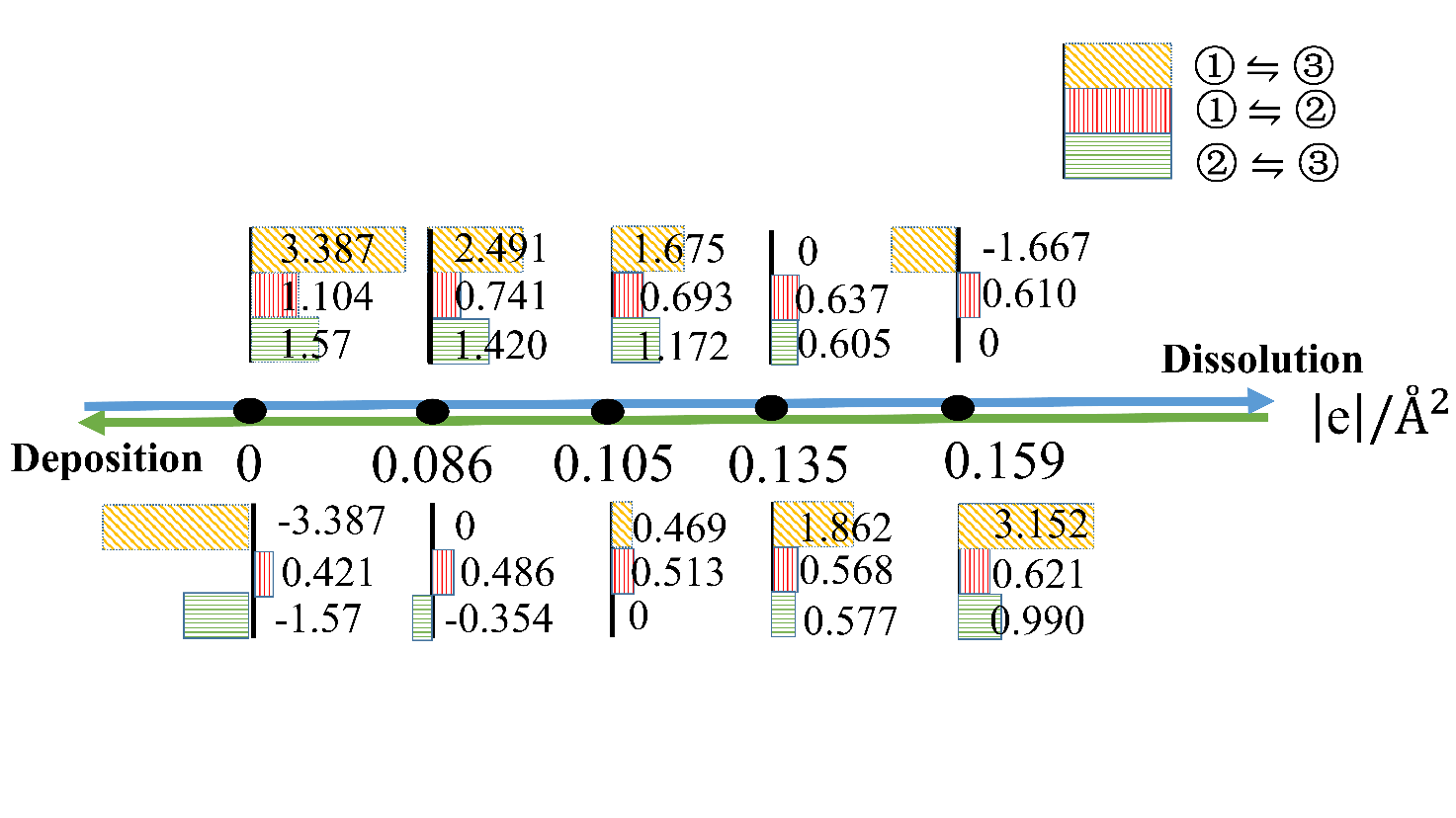
| [1] |
Koch G, Varney J, Thompson N, Moghissi O, Gould M, Payer J. International measures of prevention, application, and economics of corrosion technologies study[M]. USA: NACE International, 2016: 2-3.
|
| [2] |
Hou B R, Li X G, Ma X M, Du C W, Zhang D W, Zheng M, Xu W C, Lu D Z, Ma F B. The cost of corrosion in china[J]. NPJ Mater. Degrad., 2017, 1(7579): 29-33.
|
| [3] |
Cui K. Dictionary of safety engineering titles[M]. China: Chemical Industry Press, 1995.
|
| [4] |
Huang Y C. Metal corrosion and protection principles[M]. China: Shanghai Jiao Tong University Press, 1989.
|
| [5] |
Zhang B H, Cong W B, Yang P. Electrochemical corrosion and protection of metals[M]. China: Chemical Industry Press, 2005.
|
| [6] |
Rive D L. Electrochemical theory of corrosion of zinc in acid solutions[J]. Ann. Chim. Phys., 1830, 43: 425.
|
| [7] |
Gamburg Y D, Zangari G. Theory and practice of metal electrodeposition[M]. USA: Springer Science & Business Media, 2011.
|
| [8] |
Schlesinger M, Paunovic M. Modern electroplating[M] USA: John Wiley & Sons, 2011.
|
| [9] |
Knacke O, Stranski I N. Die theorie des kristallwachstums. In: Ergebnisse der exakten naturwissenschaften[M]. USA: Springer, 1952. 383-427.
|
| [10] |
Lorenz W. Kristallisationsüberspannungen[J]. Z. Naturfors. Sect. A-J. Phys. Sci., 1954, 9(9): 716-724.
|
| [11] |
Lorenz W. Oszillographische Überspannungsmessungen. I[J]. Z. Phys. Chemie-Int. J. Res. Phys. Chem. Chem. Phys., 1954, 58(10): 912-918.
|
| [12] |
Plieth W. The concept of dwell times in a kinetic model of alloy deposition[J]. Z. Phys. Chemie-Int. J. Res. Phys. Chem. Chem. Phys., 2003, 217(4): 383-394.
|
| [13] |
Conway B E, Bockris J O M. The mechanism of electrolytic metal deposition[J]. Proc. R. Soc. London Ser. A-Math. Phys. Eng. Sci., 1958, 248(1254): 394-403.
|
| [14] |
Vitanov T, Popov A, Budevski E. Mechanism of electrocrystallization[J]. J. Electrochem. Soc., 1974, 121(2): 207.
doi: 10.1149/1.2401782
URL
|
| [15] |
Zha Q X. Introduction to kinetics of electrode process[M]. Beijing: Science Press, 2002.
|
| [16] |
Groenenboom M C, Moffat T P, Schwarz K A. Halide-induced step faceting and dissolution energetics from atomistic machine learned potentials on Cu (100)[J]. J. Phys. Chem. C, 2020, 124(23): 12359-12369.
doi: 10.1021/acs.jpcc.0c00683
URL
|
| [17] |
Sundararaman R, Letchworth-Weaver K, Schwarz K A, Gunceler D, Ozhabes Y, Arias T. Jdftx: Software for joint density-functional theory[J]. SoftwareX, 2017, 6: 278-284.
doi: 10.1016/j.softx.2017.10.006
pmid: 29892692
|
| [18] |
Letchworth-Weaver K, Arias T. Joint density functional theory of the electrode-electrolyte interface: Application to fixed electrode potentials, interfacial capacitances, and potentials of zero charge[J]. Phys. Rev. B, 2012, 86(7): 075140.
doi: 10.1103/PhysRevB.86.075140
URL
|
| [19] |
Petrosyan S, Rigos A, Arias T. Joint density-functional theory: Ab initio study of Cr2O3 surface chemistry in solution[J]. J. Phys. Chem. B, 2005, 109(32): 15436-15444.
doi: 10.1021/jp044822k
URL
|
| [20] |
Jiang H, Sun S, Zhang T Y. Potential and distance dependent charge transfer: Hybrid first-principles/continuum calculations[J]. Comput. Mater. Sci., 2019, 169: 109121.
doi: 10.1016/j.commatsci.2019.109121
URL
|
| [21] |
Jiang H X, Sun Y Z, Sun S, Zhang T Y. Dissolving mechanisms of zinc single crystal electrode under couplings of strain and electrode potential by multiscale calculationsand symbolic regression[J]. Shanghai Metals, 2020, 42(1): 104-110.
|
| [22] |
Bronsted J. Acid and basic catalysis[J]. Chem. Rev., 1928, 5(3): 231-338.
doi: 10.1021/cr60019a001
URL
|
| [23] |
Greeley J, Mavrikakis M. Alloy catalysts designed from first principles[J]. Nat. Mater., 2004, 3(11): 810-815.
pmid: 15502837
|
| [24] |
Xu Y, Ruban A V, Mavrikakis M. Adsorption and dissociation of O2 on Pt-Co and Pt-Fe alloys[J]. J. Am. Chem. Soc., 2004, 126(14): 4717-4725.
doi: 10.1021/ja031701+
URL
|
| [25] |
Nilekar A U, Greeley J, Mavrikakis M. A simple rule of thumb for diffusion on transition-metal surfaces[J]. Angew. Chem. Int. Ed., 2006, 118(42): 7204-7207.
doi: 10.1002/ange.v118:42
URL
|
| [26] |
Gomes J R, Viñes F, Illas F, Fajín J L. Implicit solvent effects in the determination of brønstedevans-polanyi relationships for heterogeneously catalyzed reactions[J]. Phys. Chem. Chem. Phys., 2019, 21(32): 17687-17695.
doi: 10.1039/c9cp02817j
pmid: 31364629
|
| [27] |
Sundararaman R, Schwarz K A, Letchworth-Weaver K, Arias T. Spicing up continuum solvation models with salsa: The spherically averaged liquid susceptibility ansatz[J]. The J. Chem. Phys., 2015, 142(5): 054102.
doi: 10.1063/1.4906828
URL
|
| [28] |
Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Phys. Rev. Lett., 1996, 77(18): 3865.
doi: 10.1103/PhysRevLett.77.3865
pmid: 10062328
|
| [29] |
Seebauer E, Allen C. Estimating surface diffusion coefficients[J]. Prog. Surf. Sci., 1995, 49(3): 265-330.
doi: 10.1016/0079-6816(95)00039-2
URL
|

 ), 张统一a,*(
), 张统一a,*(